
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Effects of tumor microenvironment acidification on progression of pancreatic ductal adenocarcinoma: A review
Manh Tien Tran11Department of Dental Pharmacology, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama 700-8525, Japan. Correspondence: Manh Tien Tran (Department of Dental Pharmacology, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, 2-5-1 Shikata-cho, Kita-ku, Okayama, 700-8525, Japan; E-mail: trantienmanh1508@gmail.com).
Asia-Pacific Journal of Oncology 2021, 2: 27-34. https://doi.org/10.32948/ajo.2021.08.14
Received: 08 Jul 2021 | Accepted: 14 Aug 2021 | Published online: 05 Sep 2021
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive and devastating disease, which is characterized by invasiveness, rapid progression and profound resistance to treatment. It has been best characterized that tumor microenvironment such as hypoxia and nutrient deprivation contributes to cancer progression; however, the role of tumor microenvironment acidification (TMA), a major feature of tumor tissue, has not been intensively studied. Interestingly, clinicopathological clues have recently unraveled that TMA is involved in promoting cancer progression although the exact signaling pathways is poorly understood. In PDAC, the TAM is tightly regulated by proton (H+) transporters and pumps. This review dissects and summarizes the roles of these H+-extruding regulators in facilitating PDAC progression.
Key words H+ transporters, H+ pumps, TMA, PDAC
The pancreas is a complicated organ consisted of both exocrine glands (acinar and ductal cells secreting digestive enzymes into the intestinal lumen) and endocrine (α, β, δ, ε) glands, also known as the islets of Langerhans, which are responsible for secreting hormones into the blood stream [7]. Under certain extracellular stimuli such as tissue damage, stress conditions, or inflammatory factors, acinar cells can transdifferentiate into cells expressing specific ductal markers [8-10]. During acinar-to-ductal metaplasia (ADM), acinar cells acquire ‘progenitor cell-like’ properties that render them more susceptible to pro-oncogenic hits, such as activating mutations in the proto-oncogene Kirsten rat sarcoma virus (KRAS), eventually transforming them into pancreatic intra-epithelial neoplasias (PanINs). This transformation is generally considered as the initial step in PDAC development followed by sequential progression involving genetic hits in several tumor suppressor genes. Many studies have indicated that the gene encoding the proto-oncogenic GTPase KRAS as well as several tumor suppressor genes, consisting of tumor suppressor p53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), and mothers against decapentaplegic homologue 4 (SMAD4), exhibit the most frequent alterations and/or mutations in PDAC [11, 12]. Besides, RAC-beta serine/threonine-protein kinase (AKT2) is frequently overexpressed [13, 14], and the activity of its upstream regulator phosphoinositide 3-kinase (PI3K) is often enhanced in PDAC, which leads to increased cancer cell survival [15, 16].
The PDAC tumor microenvironment (TME) is principally consisted of various cell types such as fibroblasts, endothelial cells, neurons, and infiltrating immune cells as well as the extracellular matrix (ECM) proteins such as cytokines, growth factors and blood vessels. The majority of the PDAC histology is desmoplasia derived from the stroma/desmoplastic reactions. The interactions amongst components in TME and the cancer cells play a central role in facilitating immune escape, tumor progression, and metastasis. The major characteristics of TME include hypoxia, nutrient deprivation and extracellular acidification that are thought to be the potential activators of cancer progression and metastasis. Whereas hypoxia and nutrient deprivation have been well-documented, TMA in PDAC has not been intensively studied. However, recent studies have confirmed that TAM is involved in initiating the early events of malignant transformation [17-20]; more crucially, promoting tumor progression and metastasis. In the context of TMA-mediated cancer progression, it is well known that invasiveness and metastasis of cancer cells are accelerated by a variety of extracellular proteases such as metalloproteinases (MMPs), thiol proteases, serine proteases and acid proteases, which are responsible for degrading the tumor barriers, creating ideal condition to favor tumor metastasis. Notably, the proteolytic activity of these enzymes could be optimized in the TMA.
Cancer cells exert aerobic glycolysis to generate energy and supply intermediates for macromolecule biosynthetic that are required for cell survival, differentiation and proliferation [21]. A common by-product generated by this metabolic pathway is lactate, which is converted from pyruvate by lactic acid dehydrogenase-A (LDHA) [22]. The cytosolically accumulated lactate is extruded into the TME via activated H+-linked monocarboxylate transporters (MCTs), subsequently acidifying TME [23]. Lactate activates vascular endothelial growth factor (VEGF) [24], transforming growth factor beta (TGFβ) [25], interleukin-1 (IL-1) [26] and HIF-1 [27]. Also, it is worth noting that glycolytic inactivation in cancer cells still facilitates acidification of the TME [28-30], indicating that the H+ efflux pathways might be regulated by other H+ extruders, in addition to MCTs, which include Na+/H+ exchangers (NHEs) [31, 32], sodium/bicarbonate transporters (NBCs) [33], V-type H+ ATPases [34, 35], and carbonic anhydrases (CAs) [36]. Therefore, addressing the question of why cancer cells, but not normal, non-transformed cells, can thrive in the TME is of utmost importance since therapies interfering with the TME might provide useful clinical strategies for patients with cancer. This review will summarize the specific roles of H+ transporters, pumps and channels in facilitating the PDAC development and progression.
Alternatively, lactate discharging might be mediated through gap junctions that are connected by connexin channels. Previously Dovmark et al. demonstrated that Cx43 channels played a role as the crucial conduits for transmitting lactate from glycolytic PDAC cells into the neighboring cells, which triggers alkalization of recipient cells [47]. Markedly, cell-cell contact via this junctional flux is thermodynamically insensitive to TME, which possibly compensates for the TME-induced reduction of MCT activity. PDAC tumors are characterized by profoundly under-perfused regions requiring efficient mechanisms for discharging and transmitting lactate across clusters of PDAC cells [47], towards specific regions that possess the most favorable transmembrane gradient for MCT-assisted off-loading lactate. Due to it, the junctional flux of lactate benefits tumor growth through (1) providing an extracellular pH (pHe)-insensitive route for discharging metabolic wastes from glycolytic PDAC cells, and (2) alkalizing neighboring cells such as fibroblasts. It is suggested that blocking Cx43 channels by the specific blocker such as inoxynil and ioxynil octanoate might be a promising strategy to inhibit PDAC progression. However, more in vivo experiments using animal models are needed to test the anti-cancer efficaciousness of these specific blockers of Cx43 channels.
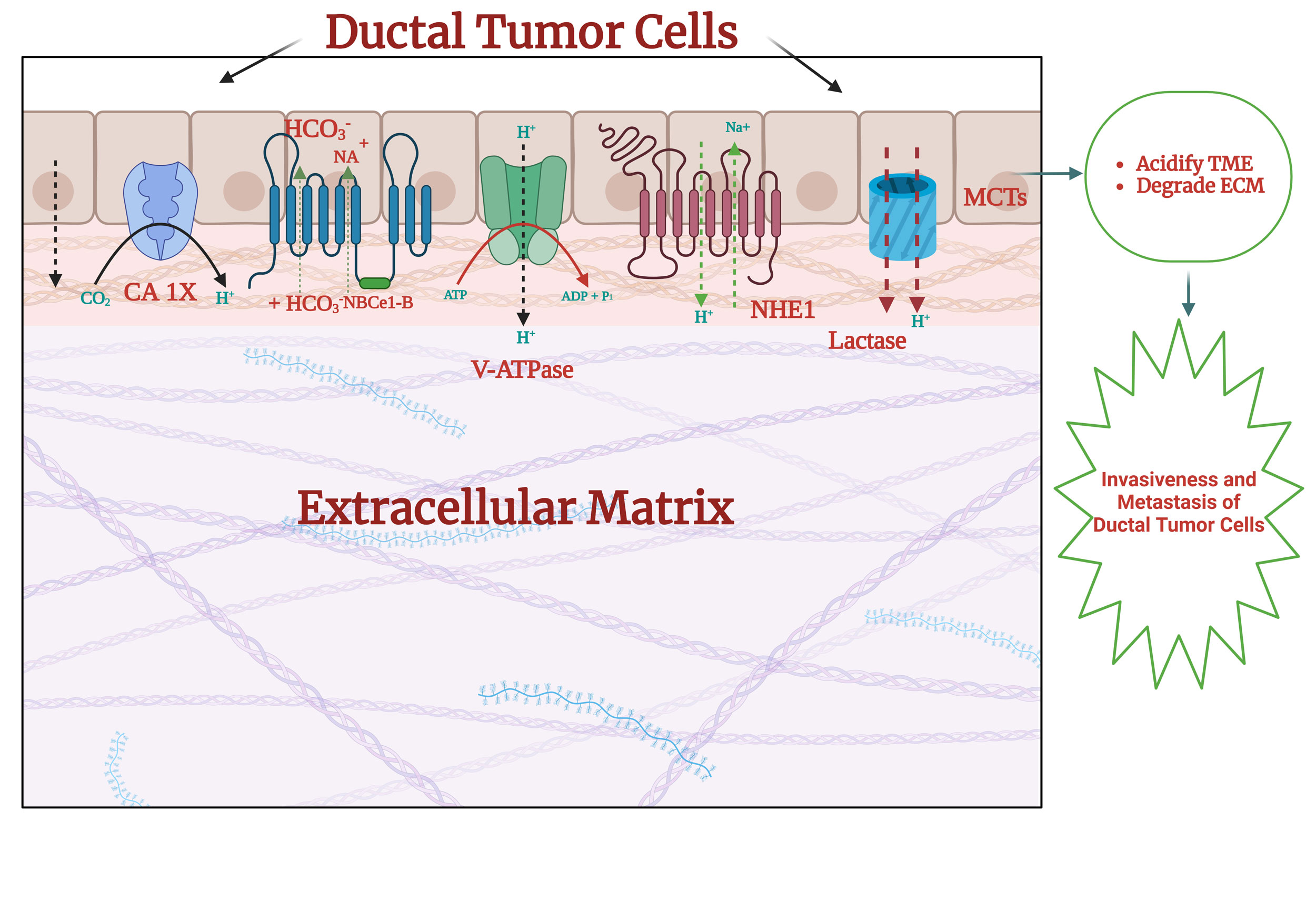 Figure 1. The possible mechanism underlying the TMA-mediated regulation
of invasiveness and metastasis. The anaerobic glycolysis of ductal tumor
cells enhances lactate production. Then, lactate is transported across
plasma membrane via MCTs, mainly MCT-1 and -4 in a H+-dependent manner.
In PDAC cells, NHE1, V-ATPases, and CA IX are frequently overexpressed
to enhance the H+ transport into extracellular milieu, contributing to
acidify the TME. Acidification of the TME subsequently provides the
ideal milieu for enzymatic activity of MMPs to degrade the ECM, thereby
facilitating the invasiveness and metastasis of PDAC cells.
Figure 1. The possible mechanism underlying the TMA-mediated regulation
of invasiveness and metastasis. The anaerobic glycolysis of ductal tumor
cells enhances lactate production. Then, lactate is transported across
plasma membrane via MCTs, mainly MCT-1 and -4 in a H+-dependent manner.
In PDAC cells, NHE1, V-ATPases, and CA IX are frequently overexpressed
to enhance the H+ transport into extracellular milieu, contributing to
acidify the TME. Acidification of the TME subsequently provides the
ideal milieu for enzymatic activity of MMPs to degrade the ECM, thereby
facilitating the invasiveness and metastasis of PDAC cells.
Besides, other NHE isoforms, such as NHE6 and NHE9, have also been confirmed to be essential for tumor pH regulation, carcinogenesis, and development of chemoresistance [55, 56]. NHE7 localized in the trans-Golgi network (TGN) played a role in acidifying the TGN and controlling pHi in PDAC cells [57]. NHE7 was up-regulated in PDAC tumors, and correlated with poor prognosis and patient survival [58].
Additionally, conversion of CO2 to HCO3- and H+ is catalyzed by CAs that are the transmembrane zinc metallo-enzymes. In mammals, there are five cytosolic forms (CA I, CA II, CA III, CA VII, and CA XIII), five membrane-bound enzymes (CA IV, CA IX, CA XII, CA XIV, and CA XV), two mitochondrial forms (CA VA and CA VB), and a secreted CA isozyme (CA VI) [64]. Among CA isoforms, CA IX is best characterized to be critical for regulating pHi [65] and acidifying extracellular environment [66]. CA IX is tightly associated with promotion of the aggressive/invasive phenotype of tumors [67]; importantly, it was identified to be a prominent biomarker of poor patient prognosis for many solid cancers [68]. CA IX that was activated by the HIF-1α pathways under hypoxic conditions played an important role in maintaining the hypoxic tumor microenvironment, which promoted tumor growth [68]. Structurally, CA IX consisting of an N-terminal proteoglycan-like domain, a CA domain, a transmembrane anchor that is associated with plasma membrane via a single-pass transmembrane region, and a C-terminal cytoplasmic tail, is responsible for ameliorating the CO2 hydration as well as accelerating CO2 diffusion and H+ mobility in the tumor tissue. Moreover, it was demonstrated that CA IX spatially and functionally cooperated with a variety of acid extruders and HCO3- transporters such as NBCe1-B, NBCn1 [69-71], and/or lactate and H+-exporting MCT1, MCT4, and NHE1 [72]. Recently, CA IX has recently been identified to be a pro-migratory factor facilitating cell movement and invasion by weakening intercellular adhesion and increasing cell dissociation via alleviating E-cadherin binding to β-catenin [70, 73]. In PDAC, McDonald et al. revealed an important role of CA IX in mediating the survival of pancreatic cancer cells by modulating pHi and glycolysis under hypoxic conditions [74]. Furthermore, Yuji Li et al. reported that knockdown of CA IX expression markedly inhibited the invasiveness and metastasis of PDAC cells lines (AsPC-1 and Miacapa), suggesting a pro-tumorigenic role of CA IX in initiating the PDAC progression [75]. Together, blocking the functional roles of CA IX might be an up-and-coming solution to alleviate the PDAC development and progression.
In the context of cancer, plasma membrane V-ATPases are necessary to maintain not only an alkaline intracellular environment that is favorable for cancer cell growth, but also an acidic extracellular environment that favors cancer cell invasion [84]. Specifically, the elevated expression levels of plasma membrane V-ATPases were found in metastatic breast cancer cells [85], and blocking V-ATPases by specific inhibitors such as bafilomycin and concanamycin A (ConA) diminished the invasiveness of these cells in a manner proposed to involve plasma membrane-localized V-ATPases. Interestingly, the similar effects of blockade of V-ATPase activity were observed in melanoma cells [34] and prostate cancer cells [86, 87].
V-ATPase function is of particular interest in PDAC, given the reliance of this exceptionally aggressive cancer on nutrient scavenging, and increased lysosomal catabolism, processes critically dependent on V-ATPase activity. In PDAC patient tissues, expression levels of V-ATPase subunits V1E and V0c were correlated with cancer stage. Nonetheless, contrary to what was found in other cancer cells, V-ATPase inhibition did not consistently weaken the invasiveness of PDAC cells. Concanamycin-induced blockade of V-ATPase activity enhanced MMP2 activity, but weakened MMP9 activity [88]; however, in general, the inhibition of V-ATPase activity alleviated the invasiveness of PDAC cells [88]. Furthermore, V-ATPase activity is confirmed to be essential for degrading MT-MMP [89, 90], which was important for promoting the invasiveness of PDAC cells [91, 92]. Hayashi et al. previously reported that vacuolar V-ATPases was essential for regulating the transport of H+ from cytosol into endocytic organelles and secretory vesicles, which are responsible for transporting H+ through plasma membrane [93], indicating an important role of vacuolar V-ATPases in promoting extracellular acidification in PDAC cells. However, more studies are needed to reveal all aspects of functional roles of V-ATPases in regulating PDAC progression before establishing the drug program development that targets V-ATPases for treatment of patients with pancreatic cancer.
In total, I have presented the major effects of the TMA on facilitating progression of PDAC, indicating that TMA may be considered as a hallmark of PDAC. In fact, the current mechanisms underlying the TMA-mediated regulation of PDAC progression is poorly understood, and thus, more in-depth studies should be conducted to improve our knowledge of the relationship between the TMA and PDAC. This, in turn, may open a therapeutic window for pancreatic cancer treatments. In this review, the roles of H+ transporters (NHEs, NBCs, and MCTs) and pumps (V-ATPases) in facilitating the TMA by providing the optimal conditions for MMP degradation in the ECM, may play a critical role in the invasiveness of PDAC cells (Figure 1). Therefore, blockade of such H+ regulators by the specific blockers would be a promising approach that may provide a possible treatment for patients with pancreatic cancer.
Not applicable.
Ethical policy
No research involving experimentation on human or animal subjects was conducted.
Author contributions
The author contributed solely to the work.
Competing interests
The author declares no conflict of interest with the work.
- Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, et al: Pancreatic cancer. Nat Rev Dis Primers 2016, 2: 16022.
- Ilic M, Ilic I: Epidemiology of pancreatic cancer. World J Gastroenterol 2016, 22(44): 9694-9705.
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011, 364(19): 1817-1825.
- Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al: Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997, 15(6): 2403-2413.
- Kunzmann V, Siveke JT, Algül H, Goekkurt E, Siegler G, Martens U, Waldschmidt D, Pelzer U, Fuchs M, Kullmann F, et al: Nab-paclitaxel plus gemcitabine versus nab-paclitaxel plus gemcitabine followed by FOLFIRINOX induction chemotherapy in locally advanced pancreatic cancer (NEOLAP-AIO-PAK-0113): a multicentre, randomised, phase 2 trial. Lancet Gastroenterol Hepatol 2021, 6(2): 128-138.
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM: Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014, 74(11): 2913-2921.
- Gylfe E, Tengholm A: Neurotransmitter control of islet hormone pulsatility. Diabetes Obes Metab 2014, 16 Suppl 1: 102-110.
- Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt, Pan FC, Akiyama H, Wright CV, Jensen K, et al: Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22(6): 737-750.
- He P, Yang JW, Yang VW, Bialkowska AB: Krüppel-like Factor 5, Increased in Pancreatic Ductal Adenocarcinoma, Promotes Proliferation, Acinar-to-Ductal Metaplasia, Pancreatic Intraepithelial Neoplasia, and Tumor Growth in Mice. Gastroenterology 2018, 154(5): 1494-1508.e13.
- Chen NM, Singh G, Koenig A, Liou GY, Storz P, Zhang JS, Regul L, Nagarajan S, Kühnemuth B, Johnsen SA, et al: NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015, 148(5): 1024-1034.e9.
- Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, Klempner SJ, Hendifar A, Milind JM, Golan T, et al: Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156(8): 2242-2253.e4.
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al: Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518(7540): 495-501.
- Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Tanno S, Skele KL, Hoffman JP, Testa JR: Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem 2002, 87(4): 470-476.
- Semba S, Moriya T, Kimura W, Yamakawa M: Phosphorylated Akt/PKB controls cell growth and apoptosis in intraductal papillary-mucinous tumor and invasive ductal adenocarcinoma of the pancreas. Pancreas 2003, 26(3): 250-257.
- Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN, Papke B, et al: Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov 2020, 10(1): 104-123.
- Chalabi-Dchar M, Cassant-Sourdy S, Duluc C, Fanjul M, Lulka H, Samain R, Roche C, Breibach F, Delisle MB, Poupot M, et al: Loss of Somatostatin Receptor Subtype 2 Promotes Growth of KRAS-Induced Pancreatic Tumors in Mice by Activating PI3K Signaling and Overexpression of CXCL16. Gastroenterology 2015, 148(7): 1452-1465.
- Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, et al: Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 2009, 69(6): 2260-2268.
- Kondo A, Yamamoto S, Nakaki R, Shimamura T, Hamakubo T, Sakai J, Kodama T, Yoshida T, Aburatani H, Osawa T: Extracellular Acidic pH Activates the Sterol Regulatory Element-Binding Protein 2 to Promote Tumor Progression. Cell Rep 2017, 18(9): 2228-2242.
- Calorini L, Peppicelli S, Bianchini F: Extracellular acidity as favouring factor of tumor progression and metastatic dissemination. Exp Oncol 2012, 34(2): 79-84.
- Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, Aranda Lopez P, Hahlbrock J, Muth S, Endo S, et al: Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol 2018, 19(12): 1319-1329.
- Ganapathy-Kanniappan S, Geschwind JF: Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 2013, 12: 152.
- Hirschhaeuser F, Sattler UG, Mueller-Klieser W: Lactate: a metabolic key player in cancer. Cancer Res 2011, 71(22): 6921-6925.
- Hiremath SA, Surulescu C, Jamali S, Ames S, Deitmer JW, Becker HM: Modeling of pH regulation in tumor cells: Direct interaction between proton-coupled lactate transporters and cancer-associated carbonic anhydrase. Math Biosci Eng 2018, 16(1): 320-337.
- Ruan GX, Kazlauskas A: Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem 2013, 288(29): 21161-21172.
- Tu CE, Hu Y, Zhou P, Guo X, Gu C, Zhang Y, Li A, Liu S: Lactate and TGF-β antagonistically regulate inflammasome activation in the tumor microenvironment. J Cell Physiol 2021, 236(6): 4528-4537.
- Wu J, Hong Y, Wu T, Wang J, Chen X, Wang Z, Cheng B, Xia J: Stromal-epithelial lactate shuttle induced by tumor‑derived interleukin‑1β promotes cell proliferation in oral squamous cell carcinoma. Int J Mol Med 2018, 41(2): 687-696.
- De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P: Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One 2012, 7(10): e46571.
- Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK: Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res 2002, 8(4): 1284-1291.
- Yamagata M, Hasuda K, Stamato T, Tannock IF: The contribution of lactic acid to acidification of tumours: studies of variant cells lacking lactate dehydrogenase. Br J Cancer 1998, 77(11): 1726-1731.
- Newell K, Franchi A, Pouysségur J, Tannock I: Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc Natl Acad Sci U S A 1993, 90(3): 1127-1131.
- Wang H, Cai J, Du S, Wei W, Shen X: LAMC2 modulates the acidity of microenvironments to promote invasion and migration of pancreatic cancer cells via regulating AKT-dependent NHE1 activity. Exp Cell Res 2020, 391(1): 111984.
- Xie R, Wang H, Jin H, Wen G, Tuo B, Xu J: NHE1 is upregulated in gastric cancer and regulates gastric cancer cell proliferation, migration and invasion. Oncol Rep 2017, 37(3): 1451-1460.
- Li JM, Lee S, Zafar R, Shin E, Choi I: Sodium bicarbonate transporter NBCe1 regulates proliferation and viability of human prostate cancer cells LNCaP and PC3. Oncol Rep 2021, 46(1): 129.
- Nishisho T, Hata K, Nakanishi M, Morita Y, Sun-Wada GH, Wada Y, Yasui N, Yoneda T: The a3 isoform vacuolar type H⁺-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol Cancer Res 2011, 9(7): 845-855.
- McGuire C, Cotter K, Stransky L, Forgac M: Regulation of V-ATPase assembly and function of V-ATPases in tumor cell invasiveness. Biochim Biophys Acta 2016, 1857(8): 1213-1218.
- Pastorekova S, Gillies RJ: The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev 2019, 38(1-2): 65-77.
- Ancey PB, Contat C, Meylan E: Glucose transporters in cancer - from tumor cells to the tumor microenvironment. Febs j 2018, 285(16): 2926-2943.
- Wu DH, Liang H, Lu SN, Wang H, Su ZL, Zhang L, Ma JQ, Guo M, Tai S, Yu S: miR-124 Suppresses Pancreatic Ductal Adenocarcinoma Growth by Regulating Monocarboxylate Transporter 1-Mediated Cancer Lactate Metabolism. Cell Physiol Biochem 2018, 50(3): 924-935.
- Halestrap AP: The SLC16 gene family - structure, role and regulation in health and disease. Mol Aspects Med 2013, 34(2-3): 337-349.
- Sukeda A, Nakamura Y, Nishida Y, Kojima M, Gotohda N, Akimoto T, Ochiai A: Expression of Monocarboxylate Transporter 1 Is Associated With Better Prognosis and Reduced Nodal Metastasis in Pancreatic Ductal Adenocarcinoma. Pancreas 2019, 48(8): 1102-1110.
- Wu DH, Liang H, Lu SN, Wang H, Su ZL, Zhang L, Ma JQ, Guo M, Tai S, Yu S: miR-124 Suppresses Pancreatic Ductal Adenocarcinoma Growth by Regulating Monocarboxylate Transporter 1-Mediated Cancer Lactate Metabolism. Cell Physiol Biochem 2018, 50(3): 924-935.
- Schneiderhan W, Scheler M, Holzmann KH, Marx M, Gschwend JE, Bucholz M, Gress TM, Seufferlein T, Adler G, Oswald F: CD147 silencing inhibits lactate transport and reduces malignant potential of pancreatic cancer cells in in vivo and in vitro models. Gut 2009, 58(10): 1391-1398.
- Xu J, Lu Y, Qiu S, Chen ZN, Fan Z: A novel role of EMMPRIN/CD147 in transformation of quiescent fibroblasts to cancer-associated fibroblasts by breast cancer cells. Cancer Lett 2013, 335(2): 380-386.
- Sun J, Hemler ME: Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res 2001, 61(5): 2276-2281.
- Ganor Y, Grinberg I, Reis A, Cooper I, Goldstein RS, Levite M: Human T-leukemia and T-lymphoma express glutamate receptor AMPA GluR3, and the neurotransmitter glutamate elevates the cancer-related matrix-metalloproteinases inducer CD147/EMMPRIN, MMP-9 secretion and engraftment of T-leukemia in vivo. Leuk Lymphoma 2009, 50(6): 985-997.
- Tang J, Guo YS, Yu XL, Huang W, Zheng M, Zhou YH, Nan G, Wang JC, Yang HJ, Yu JM, et al: CD147 reinforces [Ca2+]i oscillations and promotes oncogenic progression in hepatocellular carcinoma. Oncotarget 2015, 6(33): 34831-34845.
- Dovmark TH, Saccomano M, Hulikova A, Alves F, Swietach P: Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene 2017, 36(32): 4538-4550.
- Slepkov E, Fliegel L: Structure and function of the NHE1 isoform of the Na+/H+ exchanger. Biochem Cell Biol 2002, 80(5): 499-508.
- Wang J, Zhou Y, Ma L, Cao S, Gao W, Xiong Q, Wang K, Yang L: CIAPIN1 Targeted NHE1 and ERK1/2 to Suppress NSCLC Cells' Metastasis and Predicted Good Prognosis in NSCLC Patients Receiving Pulmonectomy. Oxid Med Cell Longev 2019, 2019: 1970818.
- Beltrán AR, Ramírez MA, Carraro-Lacroix LR, Hiraki Y, Rebouças NA, Malnic G: NHE1, NHE2, and NHE4 contribute to regulation of cell pH in T84 colon cancer cells. Pflugers Arch 2008, 455(5): 799-810.
- Wu Y, Zhang M, Liu R, Zhao C: Oxidative Stress-Activated NHE1 Is Involved in High Glucose-Induced Apoptosis in Renal Tubular Epithelial Cells. Yonsei Med J 2016, 57(5): 1252-1259.
- Frantz C, Karydis A, Nalbant P, Hahn KM, Barber DL: Positive feedback between Cdc42 activity and H+ efflux by the Na-H exchanger NHE1 for polarity of migrating cells. J Cell Biol 2007, 179(3): 403-410.
- Olszewski U, Hlozek M, Hamilton G: Activation of Na+/H+ exchanger 1 by neurotensin signaling in pancreatic cancer cell lines. Biochem Biophys Res Commun 2010, 393(3): 414-419.
- Cardone RA, Greco MR, Zeeberg K, Zaccagnino A, Saccomano M, Bellizzi A, Bruns P, Menga M, Pilarsky C, Schwab A, et al: A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 2015, 17(2): 155-166.
- Lucien F, Pelletier PP, Lavoie RR, Lacroix JM, Roy S, Parent JL, Arsenault D, Harper K, Dubois CM: Hypoxia-induced mobilization of NHE6 to the plasma membrane triggers endosome hyperacidification and chemoresistance. Nat Commun 2017, 8: 15884.
- Llongueras J, Kondapalli K, Hack A, Capilla‐González V, Smith C, Guerrero‐Cazares H, Quiñones‐Hinojosa A, Rao R: Na+/H+ exchanger NHE9 affects tumor progression of human glioblastomas by altering endosomal pH (893.34). The FASEB J 2014, 28: 893.834.
- Galenkamp KMO, Sosicka P, Jung M, Recouvreux MV, Zhang Y, Moldenhauer MR, Brandi G, Freeze HH, Commisso C: Golgi Acidification by NHE7 Regulates Cytosolic pH Homeostasis in Pancreatic Cancer Cells. Cancer Discov 2020, 10(6): 822-835.
- Parker MD, Boron WF: The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol Rev 2013, 93(2): 803-959.
- Boedtkjer E, Moreira JM, Mele M, Vahl P, Wielenga VT, Christiansen PM, Jensen VE, Pedersen SF, Aalkjaer C: Contribution of Na+,HCO3(-)-cotransport to cellular pH control in human breast cancer: a role for the breast cancer susceptibility locus NBCn1 (SLC4A7). Int J Cancer 2013, 132(6): 1288-1299.
- Parks SK, Pouyssegur J: The Na(+)/HCO3(-) Co-Transporter SLC4A4 Plays a Role in Growth and Migration of Colon and Breast Cancer Cells. J Cell Physiol 2015, 230(8): 1954-1963.
- Lee S, Axelsen TV, Jessen N, Pedersen SF, Vahl P, Boedtkjer E: Na(+),HCO(3)(-)-cotransporter NBCn1 (Slc4a7) accelerates ErbB2-induced breast cancer development and tumor growth in mice. Oncogene 2018, 37(41): 5569-5584.
- Wong P, Kleemann HW, Tannock IF: Cytostatic potential of novel agents that inhibit the regulation of intracellular pH. Br J Cancer 2002, 87(2): 238-245.
- Supuran CT: Carbonic anhydrases as drug targets--an overview. Curr Top Med Chem 2007, 7(9): 825-833.
- Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J: Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res 2009, 69(1): 358-368.
- Svastová E, Hulíková A, Rafajová M, Zat'ovicová M, Gibadulinová A, Casini A, Cecchi A, Scozzafava A, Supuran CT, Pastorek J, et al: Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004, 577(3): 439-445.
- Benej M, Pastorekova S, Pastorek J: Carbonic anhydrase IX: regulation and role in cancer. Subcell Biochem 2014, 75: 199-219.
- Ivanov S, Liao SY, Ivanova A, Danilkovitch-Miagkova A, Tarasova N, Weirich G, Merrill MJ, Proescholdt MA, Oldfield EH, Lee J, et al: Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol 2001, 158(3): 905-919.
- Orlowski A, De Giusti VC, Morgan PE, Aiello EA, Alvarez BV: Binding of carbonic anhydrase IX to extracellular loop 4 of the NBCe1 Na+/HCO3- cotransporter enhances NBCe1-mediated HCO3- influx in the rat heart. Am J Physiol Cell Physiol 2012, 303(1): C69-80.
- Svastova E, Witarski W, Csaderova L, Kosik I, Skvarkova L, Hulikova A, Zatovicova M, Barathova M, Kopacek J, Pastorek J, et al: Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J Biol Chem 2012, 287(5): 3392-3402.
- Lee S, Axelsen TV, Andersen AP, Vahl P, Pedersen SF, Boedtkjer E: Disrupting Na⁺, HCO₃⁻-cotransporter NBCn1 (Slc4a7) delays murine breast cancer development. Oncogene 2016, 35(16): 2112-2122.
- Jamali S, Klier M, Ames S, Barros LF, McKenna R, Deitmer JW, Becker HM: Hypoxia-induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non-catalytic function. Sci Rep 2015, 5: 13605.
- Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, Pastorekova S: Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol 2014, 4: 400.
- McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, Nemirovsky O, Gillespie JA, Karasinska JM, Kalloger SE, et al: Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia. Gastroenterology 2019, 157(3): 823-837.
- Li Y, Dong M, Sheng W, Huang L: Roles of Carbonic Anhydrase IX in Development of Pancreatic Cancer. Pathol Oncol Res 2016, 22(2): 277-286.
- Ramirez C, Hauser AD, Vucic EA, Bar-Sagi D: Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 2019, 576(7787): 477-481.
- Sun-Wada GH, Wada Y: Role of vacuolar-type proton ATPase in signal transduction. Biochim Biophys Acta 2015, 1847(10): 1166-1172.
- Jefferies KC, Cipriano DJ, Forgac M: Function, structure and regulation of the vacuolar (H+)-ATPases. Arch Biochem Biophys 2008, 476(1): 33-42.
- Futai M, Sun-Wada GH, Wada Y, Matsumoto N, Nakanishi-Matsui M: Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc Jpn Acad Ser B Phys Biol Sci 2019, 95(6): 261-277.
- Williamson WR, Hiesinger PR: On the role of v-ATPase V0a1-dependent degradation in Alzheimer disease. Commun Integr Biol 2010, 3(6): 604-607.
- O'Callaghan KM, Ayllon V, O'Keeffe J, Wang Y, Cox OT, Loughran G, Forgac M, O'Connor R: Heme-binding protein HRG-1 is induced by insulin-like growth factor I and associates with the vacuolar H+-ATPase to control endosomal pH and receptor trafficking. J Biol Chem 2010, 285(1): 381-391.
- Strasser B, Iwaszkiewicz J, Michielin O, Mayer A: The V-ATPase proteolipid cylinder promotes the lipid-mixing stage of SNARE-dependent fusion of yeast vacuoles. Embo j 2011, 30(20): 4126-4141.
- Nishi T, Forgac M: The vacuolar (H+)-ATPases--nature's most versatile proton pumps. Nat Rev Mol Cell Biol 2002, 3(2): 94-103.
- Sennoune SR, Martinez-Zaguilan R: Plasmalemmal vacuolar H+-ATPases in angiogenesis, diabetes and cancer. J Bioenerg Biomembr 2007, 39(5-6): 427-433.
- Cotter K, Capecci J, Sennoune S, Huss M, Maier M, Martinez-Zaguilan R, Forgac M: Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J Biol Chem 2015, 290(6): 3680-3692.
- Smith GA, Howell GJ, Phillips C, Muench SP, Ponnambalam S, Harrison MA: Extracellular and Luminal pH Regulation by Vacuolar H+-ATPase Isoform Expression and Targeting to the Plasma Membrane and Endosomes. J Biol Chem 2016, 291(16): 8500-8515.
- Michel V, Licon-Munoz Y, Trujillo K, Bisoffi M, Parra KJ: Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int J Cancer 2013, 132(2): E1-10.
- Chung C, Mader CC, Schmitz JC, Atladottir J, Fitchev P, Cornwell ML, Koleske AJ, Crawford SE, Gorelick F: The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Lab Invest 2011, 91(5): 732-743.
- Määttä M, Soini Y, Liakka A, Autio-Harmainen H: Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: implications for tumor progression and clinical prognosis. Clin Cancer Res 2000, 6(7): 2726-2734.
- Knapinska AM, Estrada CA, Fields GB: The Roles of Matrix Metalloproteinases in Pancreatic Cancer. Prog Mol Biol Transl Sci 2017, 148: 339-354.
- Jiang W, Zhang Y, Kane KT, Collins MA, Simeone DM, di Magliano MP, Nguyen KT: CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol Cancer Res 2015, 13(1): 9-15.
- Ding J, Liu Y, Lai Y: Identifying MMP14 and COL12A1 as a potential combination of prognostic biomarkers in pancreatic ductal adenocarcinoma using integrated bioinformatics analysis. PeerJ 2020, 8: e10419.
- Hayashi Y, Katayama K, Togawa T, Kimura T, Yamaguchi A: Effects of bafilomycin A1, a vacuolar type H+ ATPase inhibitor, on the thermosensitivity of a human pancreatic cancer cell line. Int J Hyperthermia 2006, 22(4): 275-285.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.