
 Submit Manuscript
Submit Manuscript
Review | Open Access
Cellular senescence and tumor dormancy at the crossroads of therapy resistance, metastasis and cancer stemness
Qurrat Ul Ain1
1 School of Pharmacy, Bandung Institute of Technology, Bandung West Java, Indonesia.
Correspondence: Qurrat Ul Ain (School of Pharmacy, Bandung Institute of Technology, Jalan Ganesa, 10, 40116, Bandung West Java, Indonesia; E-mail: Aineevirk.av@gmail.com).
Asia-Pacific Journal of Oncology 2024, 5: 112-120. https://doi.org/10.32948/ajo.2024.12.25
Received: 11 Oct 2024 | Accepted: 18 Dec 2024 | Published online: 29 Dec 2024 
Key words senescence, tumor microenvironment, epithelial-to-mesenchymal transition, senolytics, senomorphics
Emerging evidence now suggests a key role of senescence in tumor dormancy as chemotherapy-induced senescent tumor cells have been shown to escape growth arrest and regain proliferative capacity [14]. For instance, irinotecan-treated colorectal and breast cancer cells exhibited senescence markers such as β-galactosidase activity, indicating a halt in proliferation. However, a subset of these cells bypassed the senescent state, resuming division and forming viable tumors when transplanted into immunocompromised mice, highlighting the potential for therapy resistance and tumor relapse [15, 16]. This escape from senescence has been linked to loss of p53 or Suv39h1, leading to transcriptomic changes that enable aggressive tumor formation [12]. Similarly, senescent tumor cells, marked by senescent transcriptomic profiles and SASP, have been shown to repopulate the tumor after chemotherapy in both organoid systems and patient-derived contexts [17]. Mechanisms driving this escape of senescent/dormant state include the loss of cell cycle regulators, such as p53 or p16INK4a, or upregulation of cell cycle drivers like Cdk1, cyclins, and c-myc. Furthermore, genomic instability in senescent tumor cells makes them prone to acquiring such imbalances [18, 19]. However, senescent cells with intact p53/p16INK4a may also escape, implicating additional factors such as polyploidy, autophagy, metabolic shifts, and SASP-related autocrine or paracrine effects in senescence escape [20] These observations highlight senescence as a determining factor and inducer of tumor dormancy (Figure 1).
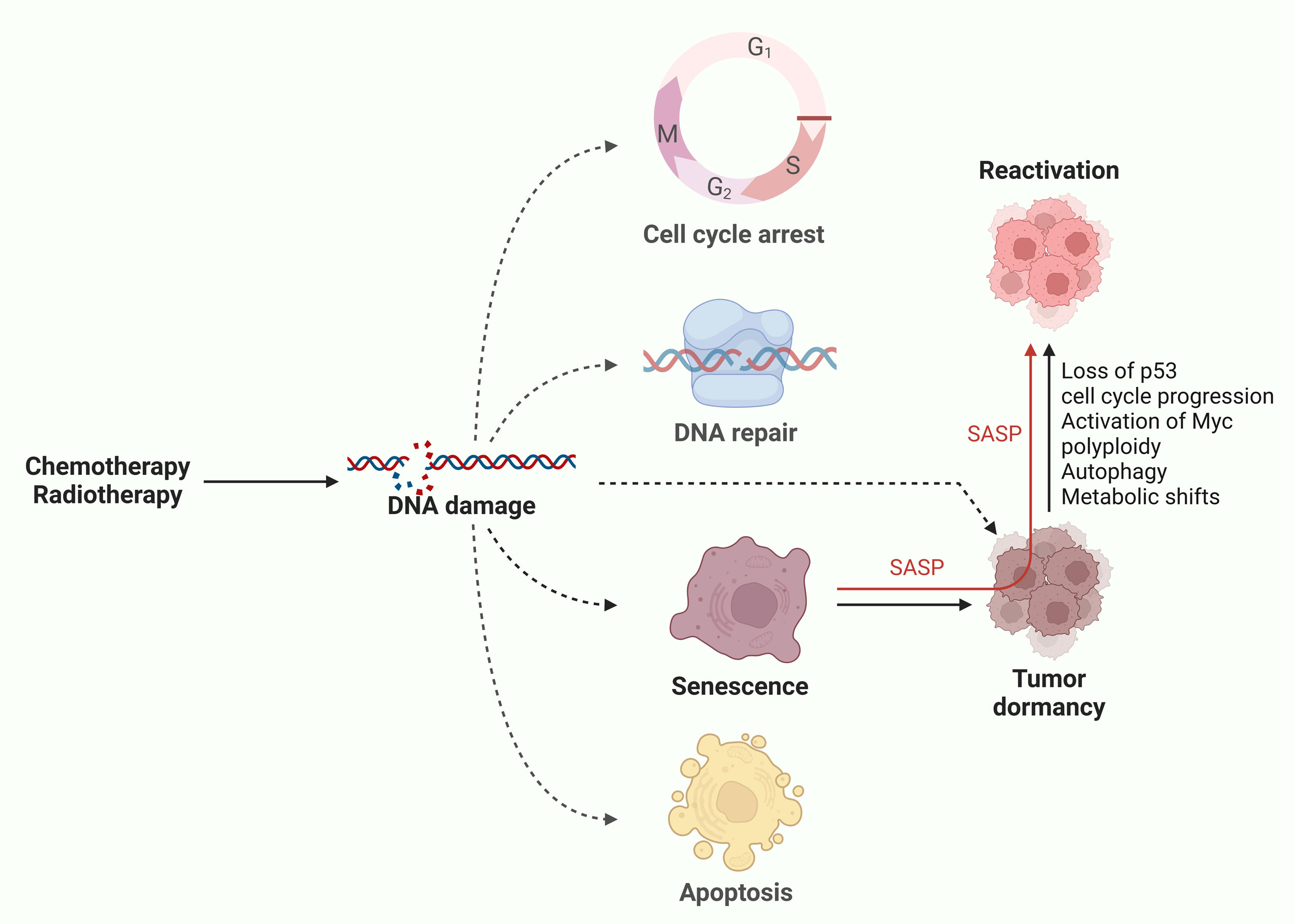 Figure 1. Cellular senescence as an inducer of tumor dormancy. Chemotherapy and radiotherapy induces DNA damage in tumor cells that leads to fate determination resulting in either of cell cycle arrest, DNA repair (revival), senescence or apoptotic cell death. Senescence can lead to tumor dormancy to evade therapeutic eradication. Upon favorable conditions, these senescent/dormant cells reactivate through plethora of mechanisms and repopulate the tumor.
Figure 1. Cellular senescence as an inducer of tumor dormancy. Chemotherapy and radiotherapy induces DNA damage in tumor cells that leads to fate determination resulting in either of cell cycle arrest, DNA repair (revival), senescence or apoptotic cell death. Senescence can lead to tumor dormancy to evade therapeutic eradication. Upon favorable conditions, these senescent/dormant cells reactivate through plethora of mechanisms and repopulate the tumor.
Therapy-induced senescence can contribute to tumor progression through the SASP, involving the secretion of pro-inflammatory cytokines (IL-6, IL-8), growth factors (VEGF, TGF-β), chemokines (CCL2, CXCL1), and proteases (MMP-3, MMP-9). Together, these factors alter the TME, creating conditions supportive of cancer cell survival, therapy resistance, immune evasion, and metastasis (Figure 2) [25, 26]. Interleukin-6 (IL-6) and IL-8 activate signaling pathways such as STAT3 and NF-κB signaling, thereby contributing to resistance to therapeutic interventions [27]. Secreted as a part of the SASP, proteases (such as MMP-9) degrade ECM components, enable cancer cells to invade surrounding tissues and evade immune surveillance [28]. Immunosuppressive cells also get recruited by SASP, such as regulatory T cells, which inhibit cytotoxic T lymphocytes and natural killer cells [29], thereby facilitating immune evasion and therapy resistance.
Therapeutic stress may induce tumor dormancy as well, through the activation of signaling pathways like TGF-β and BMP signaling, which suppress proliferation via SMAD-mediated transcriptional repression of MYC and other cell cycle regulators [30]. Dormant cells generally reside in protective niches, such as the bone marrow, where they receive survival signals from the TME [5]. Hypoxia, a key characteristic of the TME, serves as a significant factor in inducing dormancy. Under conditions of reduced oxygen availability, hypoxia-inducible factors (HIF-1α and HIF-2α) become stabilized, leading to decreased mitochondrial activity and lower energy expenditure. This adaptive response enhances cellular survival during periods of metabolic stress [31]. Hypoxia has been shown to trigger the expression of dormancy-associated genes such as NR2F1, DEC2, and p27. These genes play crucial roles in promoting cellular quiescence, enabling tumor cells to remain dormant and evade therapeutic interventions. Notably, these modifications persist after tumor cells disseminate to the lungs, rendering hypoxia-conditioned disseminated tumor cells more likely to enter a dormant state [32]. In addition to hypoxia signaling, autophagy also plays a vital role in maintaining dormancy by recycling damaged cellular components and providing essential energy during stressful conditions, such as nutrient deprivation or low oxygen often associated with hypoxia. This process helps dormant cancer cells survive in hostile environments, supporting their long-term persistence and ability to evade therapeutic treatments [33]. Dormant cancer cells, although not SASP secretors themselves, exploit SASP-modified TME to evade immune clearance. For instance, SASP-induced upregulation of PD-L1 on nearby cancer cells allows dormant cells to escape T-cell-mediated killing [34]. Collectively, dormant and senescent cells share similarities, including resistance to apoptosis and reliance on survival pathways. Both states allow cancer cells to evade therapies designed to target rapidly dividing cells, contributing to minimal residual disease and long-term therapy resistance.
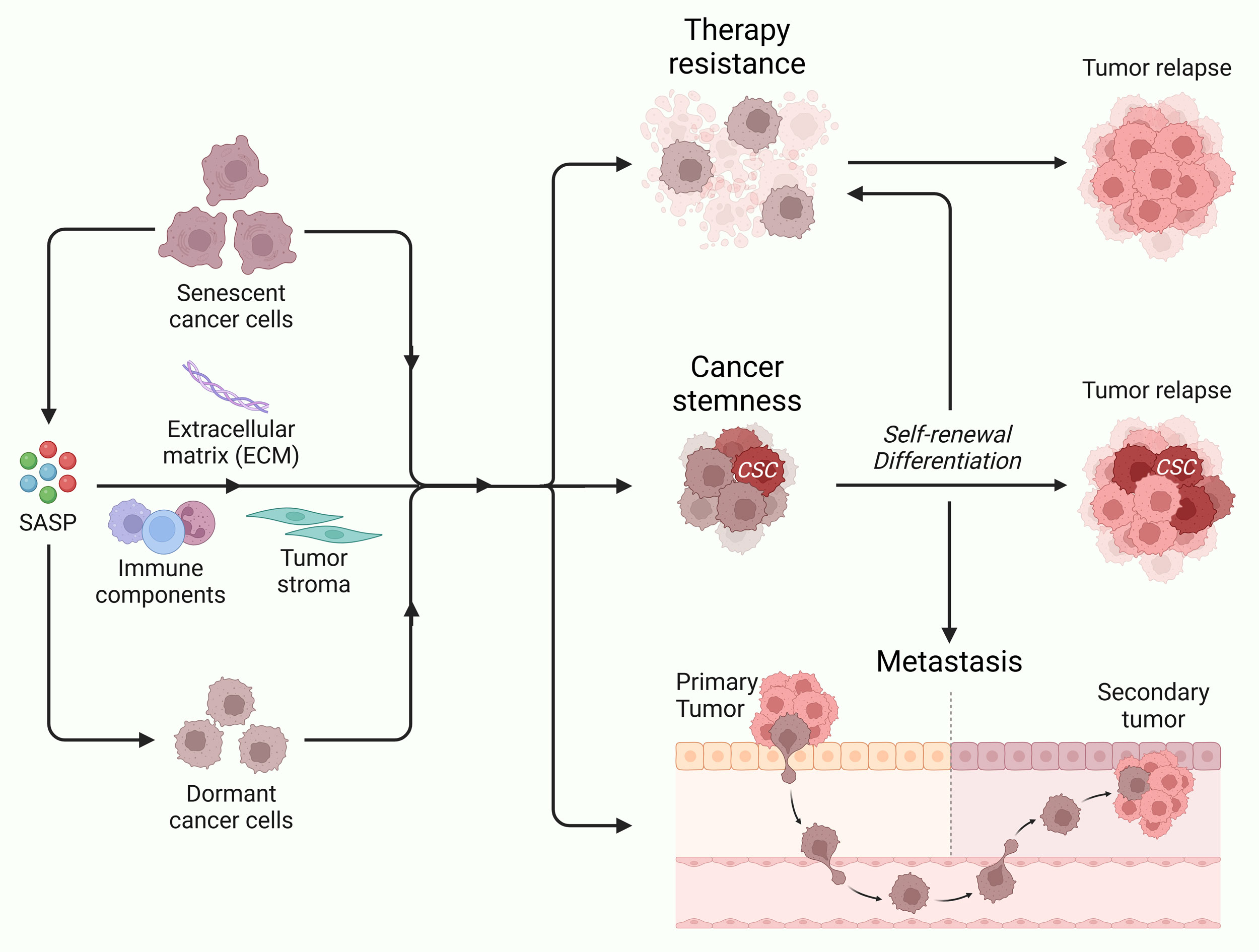 Figure 2. Cellular senescence and tumor dormancy promote therapy resistance, cancer stemness and metastasis. Through its interaction with extracellular matrix, immune cells and tumor stroma via SASP, senescent cancer promote inflammation and tumor dormancy. These events may resist repeated therapy cycles, enhance cancer stemness, and promote tumor aggressivness, leading to tumor relapse and metastasis to distant organs.
Figure 2. Cellular senescence and tumor dormancy promote therapy resistance, cancer stemness and metastasis. Through its interaction with extracellular matrix, immune cells and tumor stroma via SASP, senescent cancer promote inflammation and tumor dormancy. These events may resist repeated therapy cycles, enhance cancer stemness, and promote tumor aggressivness, leading to tumor relapse and metastasis to distant organs.
Extrinsically, senescence-driven reprogramming is mediated by the secretory activity of senescent cells in the TME. SASP factors support the maintenance and expansion of CSCs. For instance, doxorubicin- or irradiation-induced senescent cancer cells secreted SASP factors that enhanced CSC survival and growth [40]. Similarly, platinum therapies induce senescence with a SASP promoting paracrine-driven stemness in non-senescent tumor cells [41]. Notably, senescence is not confined to tumor cells; stromal cells in the TME can also adopt a senescent state upon chemotherapy exposure. These senescent stromal cells secrete SASP factors, including CXCL8, CCL20, and IL1α, which enhance chemoresistance, and stem-like transcriptomic changes in tumor cells [42]. SASP-induced EMT also correlates with the upregulation of stemness markers such as ALDH1 and CD44, reinforcing the CSC phenotype [43]. This interplay between senescence in tumor and stromal cells reinforces the CSC niche, enabling tumor dormancy and recovery after treatment. SASP can also recruit immunosuppressive cell types, including regulatory T cells, which interact with CSCs to enhance their plasticity and tumor-initiating capabilities [29]. Circulating SASP factors can remodel distant tissues, creating niches that favor CSC enrichment [40], reinforcing critical role of SASP in cancer stemness.
Dormancy serves as a protective state for CSCs, preserving their tumorigenic potential under adverse conditions. In contrast to their proliferative counterparts, dormant CSCs adopt a reversible quiescent state, allowing them to evade treatments that specifically target actively dividing cells [44]. Dormant CSCs often reside in perivascular space or bone marrow, the specialized niches, where they interact with stromal cells, endothelial cells, and ECM [45, 46]. Mediated by various signaling pathways, these interactions enforce quiescence and suppress differentiation. CXCL12-CXCR4 signaling maintains dormancy, and protects CSCs from oxidative stress by reducing mitochondrial activity [47-49]. In addition, epigenetic regulation also play a crucial role in maintaining the plasticity of dormant CSCs. Histone modifications and DNA methylation patterns in dormant cells are significantly altered to preserve their stem-like characteristics while suppressing differentiation [50]. EZH2, a component of the polycomb repressive complex, is frequently upregulated in dormant CSCs, maintaining the repression of differentiation-associated genes [51]. Hypoxia stabilizes HIF-1α, which further supports the dormant state of CSCs in the hypoxic niches by downregulating MYC and cell cycle regulators [52, 53]. Dormant CSCs’ plasticity is central to their role in cancer progression. Dormant CSCs can switch between quiescence and proliferation dynamically in response to TME cues such as inflammation, nutrient availability, or ECM remodeling [11]. VEGF-induced angiogenesis provides mitogenic signals to reactivate dormant CSCs, initiating tumor regrowth and metastasis [54]. Similarly, ECM degradation by MMPs exposes dormant CSCs to growth factors stored in the matrix, triggering reactivation and proliferation [55]. These findings highlight the potential of dormant cells in repopulating the tumor and spreading the disease by metastasizing to distant organs.
SASP can also influence non-senescent cancer cells, further underscoring its role in metastasis. Proliferative adjacent cells and/or or cell which are in close vicinity get exposed to SASP factors and acquire invasive and metastatic traits, expanding the metastatic potential of the tumor beyond the senescent population [56]. For example, IL-6-mediated STAT3 activation in non-senescent cells has been shown to increase motility and resistance to apoptosis, crucial steps for metastatic dissemination [67]. Importantly, senescence-associated inflammation is not limited to local effects. Circulating SASP factors contribute to systemic changes, such as the establishment of pre-metastatic niches in distant organs. These niches are primed with immunosuppressive cells, pro-inflammatory cytokines, and ECM remodeling enzymes, creating a fertile environment for metastatic colonization. For instance, bone marrow-derived suppressor cells and fibroblasts recruited by SASP factors enhance ECM deposition and immune evasion at secondary sites [68]. These findings highlight the dual role of SASP in promoting metastasis directly, by enabling cancer cell invasion, and indirectly, by modifying the microenvironment of both primary and secondary tumor sites.
Dormant cancer cells can act as reservoirs that enable cancer relapse and secondary tumor formation. These cells disseminate from the primary tumors early in disease progression and remain quiescent at secondary tissues for months to years [69]. As mentioned earlier, dormant cells often reside in specialized niches, such as the bone marrow, lungs, or liver, where they receive survival signals from stromal cells and the ECM [45, 46]. For example, the CXCL12-CXCR4 signaling axis between stromal cells and dormant cancer cells maintains quiescence and prevents apoptosis [70]. Hypoxia within these niches plays a critical role in maintaining dormancy. HIF-1α stabilization suppresses proliferation by downregulating MYC and other cell cycle regulators [52, 53]. Concurrently, dormant cells exhibit metabolic adaptations, such as increased reliance on oxidative phosphorylation and autophagy, which allow them to survive under nutrient-poor conditions [71]. Overexpression of CXCL12-CXCR4 axis at the downstream of NR2F1 has also been shown to reinforce quiescent dormant state in cancer cells, eventually leading to tumor recurrence and metastasis [47]. Immune evasion is another key feature of dormant cells. By downregulating MHC class I molecules, dormant cells evade detection by cytotoxic T lymphocytes [72]. They also secrete immunosuppressive cytokines, such as TGF-β, which inhibit natural killer cell activity [73, 74]. Moreover, dormant cells exploit the immunosuppressive effects of SASP in the TME, further reducing the likelihood of immune-mediated clearance. Hence, dormant cells can survive in hostile environments along with evading immune surveillance. These cell then reactivate under favorable conditions, making them formidable barriers to effective cancer treatment [75]. Reactivation of dormant cells is often triggered by changes in the TME and microenvironment at secondary sites and marks the onset of metastatic outgrowth [34]. At the primary tumor sites, inflammation, tissue remodeling, or angiogenesis can disrupt dormancy, exposing cells to mitogenic signals that re-initiate proliferation, leading to metastatic spread of the disease [76]. On the other hand, integrins, particularly β1-integrin, mediate interactions with ECM components such as fibronectin, activating focal adhesion kinase (FAK) to support exit from dormancy when cancer cell extravasate into distant organ [77]. These findings support that dormancy play key role in survival of cancer cells at primary tumor sites and distant organs where they metastasize to spread the disease.
Therapies targeting dormant cells aim to either eliminate these cells by disrupting their survival mechanisms or force them out of dormancy to sensitize them to conventional treatments. Dormant cancer cells rely on protective niches, metabolic adaptations, and survival pathways to evade apoptosis and persist in quiescence. Disrupting these mechanisms presents a viable therapeutic strategy [71]. CXCL12-CXCR4 inhibitors, such as plerixafor (AMD3100), disrupt the interaction between dormant cancer cells and their stromal niches. This strategy has shown efficacy in preclinical models of bone metastasis, where it sensitized dormant cells to chemotherapy by blocking their survival signals [87]. Similarly, integrin inhibitors targeting β1-integrin-mediated adhesion to ECM components have demonstrated potential in breaking dormancy and inducing apoptosis [88]. Autophagy inhibitors such as chloroquine and hydroxychloroquine have shown promise in disrupting the metabolic flexibility of dormant cells, rendering them more susceptible to therapy [89, 90]. Reactivation strategies aim to push dormant cells out of quiescence, making them vulnerable to standard therapies.
The immune system plays a critical role in targeting senescent and dormant cells, which often evade immune surveillance by exploiting SASP-driven immunosuppression and niche protection [91]. Immune-based therapies aim to overcome these challenges by enhancing immune recognition and clearance. Immune checkpoint inhibitors, such as anti-PD-1/PD-L1 and anti-CTLA-4 antibodies, have shown potential in reactivating T-cell-mediated immunity against senescent and dormant cancer cells [92]. By blocking PD-L1 upregulation induced by SASP or dormancy-associated pathways, Immune checkpoint inhibitors restore immune-mediated cytotoxicity. Therapeutically, the paradoxical role of senescence as a tumor-suppressor and tumor-promoter complicates targeting strategies. While eliminating senescent cells can reduce SASP-mediated stemness induction, it may also remove their tumor-suppressive functions [93], hence keeping the balance is most important. CAR-T cell therapies, engineered to target senescence- or dormancy-specific markers, represent a novel approach to eradicating resistant cancer cell populations. For example, CAR-T cells targeting uPAR, a marker associated with dormant and metastatic cells, have shown efficacy in preclinical models of breast cancer [94, 95]. Natural killer cell-based therapies are particularly effective in targeting senescent and dormant cells with low MHC-I expression. Strategies to enhance natural killer cell activity, such as IL-15 superagonists or checkpoint blockade, are under investigation for their potential to eliminate these cells [96, 97]. While therapeutic advances have shown potential in preclinical and early clinical studies, significant challenges remain in selectively targeting senescent and dormant cells without harming normal tissue homeostasis.
Identifying unique vulnerabilities in senescent and dormant cells must be key focus in future, s that, these cells can be therapeutically exploited without affecting normal tissues. In this regard, advances in single-cell technologies and spatial transcriptomics offer the potential to decode the molecular landscapes of these cells, enabling more precise therapeutic targeting [99]. Moreover, understanding and disrupting the temporal dynamics of senescence and dormancy—such as the transitions between active and quiescent states—could dictate the combination and sequential therapies to maximize efficacy [100]. In conclusion, targeting senescence and dormancy offers a great opportunity to overcome therapy resistance, cancer stemness, and reduce relapse and metastasis in cancer patients, improving long-term outcomes. Achieving this requires continued innovation, interdisciplinary research, and clinical translation of emerging therapeutic strategies, ultimately moving us closer to achieving durable cancer control.
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
QUA contributed to the conception, design, writing of this review article, figures drawing and submitted the final version of the manuscript.
Competing interests
None.
- Schmitt CA, Wang B, Demaria M: Senescence and cancer — role and therapeutic opportunities. Nat Rev Clin Oncol 2022, 19(10): 619-636.
- Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO: Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 2022, 18(10): 611-627.
- Kumari R, Jat P: Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev Biol 2021, 9: 645593.
- Santos-de-Frutos K, Djouder N: When dormancy fuels tumour relapse. Commun Biol 2021, 4(1): 747.
- Smith JT, Chai RC: Bone niches in the regulation of tumour cell dormancy. J Bone Oncol 2024, 47: 100621.
- Senft D, Jeremias I: Tumor Cell Dormancy—Triggered by the Niche. Dev Cell 2019, 49(3): 311-312.
- Lindell E, Zhong L, Zhang X: Quiescent Cancer Cells-A Potential Therapeutic Target to Overcome Tumor Resistance and Relapse. Int J Mol Sci 2023, 24(4): 3762.
- Balayan V, Guddati AK: Tumor Dormancy: Biologic and Therapeutic Implications. World J Oncol 2022, 13(1): 8-19.
- Liu Y, Lomeli I, Kron SJ: Therapy-Induced Cellular Senescence: Potentiating Tumor Elimination or Driving Cancer Resistance and Recurrence? Cells 2024, 13(15): 1281.
- Min HY, Lee HY: Cellular Dormancy in Cancer: Mechanisms and Potential Targeting Strategies. Cancer Res Treat 2023, 55(3): 720-736.
- Paul R, Dorsey JF, Fan Y: Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol Ther 2022, 231: 107985.
- Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al: Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553(7686): 96-100.
- Neophytou CM, Kyriakou TC, Papageorgis P: Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy. Int J Mol Sci 2019, 20(24): 6158.
- Saleh T, Bloukh S, Carpenter VJ, Alwohoush E, Bakeer J, Darwish S, Azab B, Gewirtz DA: Therapy-Induced Senescence: An "Old" Friend Becomes the Enemy. Cancers (Basel) 2020, 12(4): 822.
- Guillon J, Petit C, Moreau M, Toutain B, Henry C, Roché H, Bonichon-Lamichhane N, Salmon JP, Lemonnier J, Campone M, et al: Regulation of senescence escape by TSP1 and CD47 following chemotherapy treatment. Cell Death Dis 2019, 10(3): 199.
- Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, Henderson SC, Yakovlev V, Elmore LW, Toor A, et al: Tumor cell escape from therapy-induced senescence. Biochem Pharmacol 2019, 162: 202-212.
- Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, Chin CR, Durmaz C, Kawabata KC, Dhimolea E: Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer discov 2021, 11(6): 1542-1561.
- Bojko A, Czarnecka-Herok J: Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8(12): 1501.
- Hsu CH, Altschuler SJ, Wu LF: Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 2019, 178(2): 361-373.e12.
- DeLuca VJ, Saleh T: Insights into the role of senescence in tumor dormancy: mechanisms and applications. Cancer Metastasis Rev 2023, 42(1): 19-35.
- Prasanna PG, Citrin DE: Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst 2021, 113(10): 1285-1298.
- Jurkovicova D, Neophytou CM: DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int J Mol Sci 2022, 23(23): 14672.
- Dasgupta N, Arnold R, Equey A, Gandhi A, Adams PD: The role of the dynamic epigenetic landscape in senescence: orchestrating SASP expression. NPJ Aging 2024, 10(1): 48.
- Nousis L, Kanavaros P, Barbouti A: Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link? Antioxidants (Basel) 2023, 12(6): 1250.
- Chambers CR, Ritchie S, Pereira BA: Overcoming the senescence-associated secretory phenotype (SASP): a complex mechanism of resistance in the treatment of cancer. Mol Oncol 2021, 15(12): 3242-3255.
- Zhang W, Zhang K, Shi J, Qiu H, Kan C, Ma Y, Hou N, Han F: The impact of the senescent microenvironment on tumorigenesis: Insights for cancer therapy. Aging Cell 2024, 23(5): e14182.
- Dong Z, Luo Y, Yuan Z, Tian Y, Jin T, Xu F: Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol Cancer 2024, 23(1): 181.
- Liu D, Hornsby PJ: Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 2007, 67(7): 3117-3126.
- Salminen A: Immunosuppressive network promotes immunosenescence associated with aging and chronic inflammatory conditions. J Mol Med (Berl) 2021, 99(11): 1553-1569.
- Prunier C, Baker D, Ten Dijke P, Ritsma L: TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5(1): 66-78.
- Butturini E, Carcereri de Prati A, Boriero D, Mariotto S: Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int J Mol Sci 2019, 20(17): 4305.
- Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, Calvo V, Cheung JF, Bravo-Cordero JJ, Entenberg D, et al: Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol 2017, 19(2): 120-132.
- Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE: Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun 2018, 9(1): 1944.
- Zingoni A, Antonangeli F, Sozzani S, Santoni A, Cippitelli M, Soriani A: The senescence journey in cancer immunoediting. Mol Cancer 2024, 23(1): 68.
- Loh JJ, Ma S: Hallmarks of cancer stemness. Cell Stem Cell 2024, 31(5): 617-639.
- Was H, Czarnecka J, Kominek A, Barszcz K, Bernas T, Piwocka K, Kaminska B: Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol Ther 2018, 19(1): 63-75.
- Tsolou A, Lamprou I, Fortosi AO, Liousia M, Giatromanolaki A, Koukourakis MI: 'Stemness' and 'senescence' related escape pathways are dose dependent in lung cancer cells surviving post irradiation. Life Sci 2019, 232: 116562.
- Wang D, Liu L: Senescence Promotes the Recovery of Stemness among Cancer Cells via Reprograming. Biomolecules 2024, 14(3): 288.
- Roche J, Nasarre P, Gemmill R, Baldys A, Pontis J, Korch C, Guilhot J, Ait-Si-Ali S, Drabkin H: Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells. Cancers (Basel) 2013, 5(2): 334-356.
- Cahu J, Bustany S, Sola B: Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis 2012, 3(12): e446.
- Nacarelli T, Fukumoto T, Zundell JA, Fatkhutdinov N, Jean S, Cadungog MG, Borowsky ME, Zhang R: NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res 2020, 80(4): 890-900.
- Han L, Long Q, Li S, Xu Q, Zhang B, Dou X, Qian M, Jiramongkol Y: Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Res 2020, 80(16): 3383-3398.
- May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA: Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 2011, 13(1): 202.
- Francescangeli F, De Angelis ML: Dormancy, stemness, and therapy resistance: interconnected players in cancer evolution. Cancer Metastasis Rev 2023, 42(1): 197-215.
- Plaks V, Kong N, Werb Z: The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16(3): 225-238.
- Aguirre-Ghiso JA: Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007, 7(11): 834-846.
- Gao XL, Zheng M, Wang HF, Dai LL, Yu XH, Yang X, Pang X, Li L, Zhang M, Wang SS, et al: NR2F1 contributes to cancer cell dormancy, invasion and metastasis of salivary adenoid cystic carcinoma by activating CXCL12/CXCR4 pathway. BMC Cancer 2019, 19(1): 743.
- Janghorban M, Yang Y, Zhao N, Hamor C, Nguyen TM: Single-Cell Analysis Unveils the Role of the Tumor Immune Microenvironment and Notch Signaling in Dormant Minimal Residual Disease. Cancer Res 2022, 82(5): 885-899.
- Nobutani K, Shimono Y, Mizutani K, Ueda Y, Suzuki T, Kitayama M, Minami A, Momose K, Miyawaki K, Akashi K, et al: Downregulation of CXCR4 in Metastasized Breast Cancer Cells and Implication in Their Dormancy. PLoS One 2015, 10(6): e0130032.
- Robinson NJ, Parker KA, Schiemann WP: Epigenetic plasticity in metastatic dormancy: mechanisms and therapeutic implications. Ann Transl Med 2020, 8(14): 903.
- Chang CJ, Hung MC: The role of EZH2 in tumour progression. Br J Cancer 2012, 106(2): 243-247.
- Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE: HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo j 2004, 23(9): 1949-1956.
- Weston WA, Barr AR: A cell cycle centric view of tumour dormancy. Br J Cancer 2023, 129(10): 1535-1545.
- Wang L, Zhang L, Zhao L, Shao S, Ning Q, Jing X, Zhang Y, Zhao F, Liu X, Gu S, et al: VEGFA/NRP-1/GAPVD1 axis promotes progression and cancer stemness of triple-negative breast cancer by enhancing tumor cell-macrophage crosstalk. Int J Biol Sci 2024, 20(2): 446-463.
- Albrengues J, Shields MA: Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361(6409): eaao4227.
- Faggioli F, Velarde MC: Cellular Senescence, a Novel Area of Investigation for Metastatic Diseases. Cells 2023, 12(6): 860.
- Takasugi M, Yoshida Y, Ohtani N: Cellular senescence and the tumour microenvironment. Mol Oncol 2022, 16(18): 3333-3351.
- Wang B, Kohli J, Demaria M: Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6(10): 838-857.
- Niu G, Chen X: Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets 2010, 11(8): 1000-1017.
- Coppé JP, Kauser K, Campisi J, Beauséjour CM: Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 2006, 281(40): 29568-29574.
- Hsu PC, Chen YH: Interleukin-6 and Interleukin-8 Regulate STAT3 Activation Migration/Invasion and EMT in Chrysophanol-Treated Oral Cancer Cell Lines. Life (Basel) 2021, 11(5): 423.
- Palena C, Hamilton DH, Fernando RI: Influence of IL-8 on the epithelial-mesenchymal transition and the tumor microenvironment. Future Oncol 2012, 8(6): 713-722.
- Huang Y, Hong W, Wei X: The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J Hematol Oncol 2022, 15(1): 129.
- Bromberg J, Wang TC: Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009, 15(2): 79-80.
- Niland S, Riscanevo AX: Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int J Mol Sci 2021, 23(1): 146.
- Davalos AR, Coppe JP, Campisi J, Desprez PY: Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 2010, 29(2): 273-283.
- Manore SG, Doheny DL, Wong GL, Lo HW: IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front Oncol 2022, 12: 866014.
- Luo X, Fu Y, Loza AJ, Murali B, Leahy KM, Ruhland MK, Gang M, Su X, Zamani A, Shi Y, et al: Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep 2016, 14(1): 82-92.
- Tamamouna V, Pavlou E, Neophytou CM, Papageorgis P: Regulation of Metastatic Tumor Dormancy and Emerging Opportunities for Therapeutic Intervention. Int J Mol Sci 2022, 23(22): 13931.
- Yang Y, Li J, Lei W, Wang H, Ni Y, Liu Y, Yan H, Tian Y, Wang Z, Yang Z, et al: CXCL12-CXCR4/CXCR7 Axis in Cancer: from Mechanisms to Clinical Applications. Int J Biol Sci 2023, 19(11): 3341-3359.
- Pranzini E, Raugei G: Metabolic Features of Tumor Dormancy: Possible Therapeutic Strategies. Cancers (Basel) 2022, 14(3): 547.
- Goddard ET, Linde MH, Srivastava S, Klug G, Shabaneh TB, Iannone S, Grzelak CA, Marsh S, Riggio AI, Shor RE, et al: Immune evasion of dormant disseminated tumor cells is due to their scarcity and can be overcome by T cell immunotherapies. Cancer Cell 2024, 42(1): 119-134.e12.
- Ames E, Canter RJ: NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J Immunol 2015, 195(8): 4010-4019.
- Alvarez M, Dunai C: IL-2 and Anti-TGF-β Promote NK Cell Reconstitution and Anti-tumor Effects after Syngeneic Hematopoietic Stem Cell Transplantation. Cancers (Basel) 2020, 12(11): 3189.
- Baxevanis CN, Perez SA: Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State. Vaccines (Basel) 2015, 3(3): 597-619.
- Park SY, Nam JS: The force awakens: metastatic dormant cancer cells. Exp Mol Med 2020, 52(4): 569-581.
- Shibue T, Weinberg RA: Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 2009, 106(25): 10290-10295.
- Bousset L, Gil J: Targeting senescence as an anticancer therapy. Mol Cancer 2022, 16(21): 3855-3880.
- Agudo J, Aguirre-Ghiso JA, Bhatia M, Chodosh LA, Correia AL, Klein CA: Targeting cancer cell dormancy. Nat Rev Cancer 2024, 24(2): 97-104.
- von Kobbe C: Targeting senescent cells: approaches, opportunities, challenges. Aging (Albany NY) 2019, 11(24): 12844-12861.
- Skwarska A, Konopleva M: BCL-xL Targeting to Induce Apoptosis and to Eliminate Chemotherapy-Induced Senescent Tumor Cells: From Navitoclax to Platelet-Sparing BCL-xL PROTACs. Cancer Res 2023, 83(21): 3501-3503.
- Wang L, Xiong B, Lu W, Cheng Y, Zhu J, Ai G, Zhang X, Liu X, Cheng Z: Senolytic drugs dasatinib and quercetin combined with Carboplatin or Olaparib reduced the peritoneal and adipose tissue metastasis of ovarian cancer. Biomed Pharmacother 2024, 174: 116474.
- Malayaperumal S, Marotta F, Kumar MM, Somasundaram I, Ayala A, Pinto MM, Banerjee A, Pathak S: The Emerging Role of Senotherapy in Cancer: A Comprehensive Review. Clin Pract 2023, 13(4): 838-852.
- Zhou C, Huang Y, Nie S, Zhou S, Gao X, Chen G: Biological effects and mechanisms of fisetin in cancer: a promising anti-cancer agent. Eur J Med Res 2023, 28(1): 297.
- Stover DG, Gil Del Alcazar CR, Brock J, Guo H, Overmoyer B, Balko J, Xu Q, Bardia A, Tolaney SM, Gelman R, et al: Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4: 10.
- Hajimohammadebrahim-Ketabforoush M, Zali A, Shahmohammadi M, Hamidieh AA: Metformin and its potential influence on cell fate decision between apoptosis and senescence in cancer, with a special emphasis on glioblastoma. Front Oncol 2024, 14: 1455492.
- Heckmann D, Maier P, Laufs S, Wenz F, Zeller WJ, Fruehauf S, Allgayer H: CXCR4 Expression and Treatment with SDF-1α or Plerixafor Modulate Proliferation and Chemosensitivity of Colon Cancer Cells. Transl Oncol 2013, 6(2): 124-132.
- Bui T, Gu Y, Ancot F, Sanguin-Gendreau V, Zuo D, Muller WJ: Emergence of β1 integrin-deficient breast tumours from dormancy involves both inactivation of p53 and generation of a permissive tumour microenvironment. Oncogene 2022, 41(4): 527-537.
- Manic G, Obrist F, Kroemer G, Vitale I, Galluzzi L: Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol 2014, 1(1): e29911.
- Tiwari M, Srivastava P, Abbas S, Jegatheesan J, Ranjan A, Sharma S: Emerging Role of Autophagy in Governing Cellular Dormancy, Metabolic Functions, and Therapeutic Responses of Cancer Stem Cells. Cells 2024, 13(5): 447.
- Prata L, Ovsyannikova IG, Tchkonia T, Kirkland JL: Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol 2018, 40: 101275.
- Jain SS, Burton Sojo G, Sun H: The Role of Aging and Senescence in Immune Checkpoint Inhibitor Response and Toxicity. Int J Biol Sci 2024, 25(13): 7013.
- Yang J, Liu M, Hong D, Zeng M, Zhang X: The Paradoxical Role of Cellular Senescence in Cancer. Front Cell Dev Biol 2021, 9: 722205.
- Li JH, Chen YY: A Fresh Approach to Targeting Aging Cells: CAR-T Cells Enhance Senolytic Specificity. Cell Stem Cell 2020, 27(2): 192-194.
- Huang Y, Liu T: Step further towards targeted senolytic therapy: therapeutic potential of uPAR-CAR T cells for senescence-related diseases. Signal Transduct Target Ther 2020, 5(1): 155.
- Romero I, Garrido F, Garcia-Lora AM: Metastases in immune-mediated dormancy: a new opportunity for targeting cancer. Cancer Res 2014, 74(23): 6750-6757.
- Stravokefalou V, Stellas D, Karaliota S, Nagy BA, Valentin A, Bergamaschi C, Dimas K, Pavlakis GN: Heterodimeric IL-15 (hetIL-15) reduces circulating tumor cells and metastasis formation improving chemotherapy and surgery in 4T1 mouse model of TNBC. Front Immunol 2022, 13: 1014802.
- Khosla S: Senescent cells, senolytics and tissue repair: the devil may be in the dosing. Nat Aging 2023, 3(2): 139-141.
- Jin Y, Zuo Y, Li G, Liu W, Pan Y, Fan T, Fu X, Yao X, Peng Y: Advances in spatial transcriptomics and its applications in cancer research. Mol Cancer 2024, 23(1): 129.
- Truskowski K, Amend SR, Pienta KJ: Dormant cancer cells: programmed quiescence, senescence, or both? Cancer Metastasis Rev 2023, 42(1): 37-47.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.