
 Submit Manuscript
Submit Manuscript
Review | Open Access
Ubiquitin-specific proteases as emerging molecular drivers and therapeutic targets in hepatobiliary cancers
Enas Roumieh1, Waqas Bin Ismail2, Samahir Sheikh Idris3
1Department of Medicine, Faculty of Medicine, Damascus University, Damascus 11451, Syria.
2Department of Clinical Bacteriology, Parasitology and Medical Entomology Research Building, University Hospital of Heraklion, 712 01 Heraklion, Crete, Greece.
3Department of Chemical Engineering, Faculty of Engineering, Gebze Technical University, Gebze 41400, Turkey.
Correspondence: Enas Roumieh (Department of Medicine, Faculty of Medicine, Damascus university, Damascus 11451, Syria; Email: enas.roumieh@gmail.com).
Asia-Pacific Journal of Oncology 2025, 6: 55-69. https://doi.org/10.32948/ajo.2025.08.16
Received: 11 Jul 2025 | Accepted: 16 Sep 2025 | Published online: 30 Oct 2025 
Key words clinical correlations, deubiquitination, hepatobiliary cancer, inhibitors, ubiquitin-specific proteases
The main mechanism that controls protein stability and turnover, the UPS, is the focus of a quickly developing area of cancer biology. By destroying tumour suppressors or stabilising oncogenic proteins, UPS dysregulation aids in oncogenesis. By eliminating ubiquitin moieties from target proteins, deubiquitinating enzymes (DUBs) balance out ubiquitin ligases in this system [7]. The USP family is the largest and most functionally varied of the more than 100 known DUBs. According to their structural makeup, USPs are cysteine proteases that selectively cleave ubiquitin from protein substrates, influencing the processes that control immunological responses, metabolic signalling, apoptosis, DNA repair, and cell cycle progression [8, 9] (Figure 1).
Hepatobiliary malignancies highlight the increasing importance of USPs in the field of cancer biology, which has been the case for the past few years. In HCC, numerous USPs are expressed abnormally and function as molecular drivers of carcinogenesis. For example, it has been shown that USP14, which is significantly elevated in HCC tissues, promotes tumor growth through the HK2/AKT/P62 axis activation, thus linking deubiquitination to the metabolism and survival of cancer cells [10, 11]. Similarly, USP9X promotes the abnormal Wnt signaling by β-catenin's stabilization, connected with poor clinical prognosis [12]. Besides, a few others' USPs like USP7, USP10, and USP22 by regulating oncogenic transcription factors, apoptotic mediators and DNA damage repair proteins, have also been very helpful in the promotion of HCC [13, 14]. Interestingly, some USPs can inhibit tumors in a context-dependent manner, which reflects the complexity of their roles in liver cancer development.
Even though it is new, the proof for CCA is equally solid. A study recently revealed that USP21 connects the metabolic switch to the deubiquitination process by acting as a stabilizer of HSP90 and ENO1, which promote glycolysis and tumor (CCA) proliferation [15]. Another research indicates that deubiquitination of PARP1 by USP1 prevents its degradation, thus prolonging the life of CCA cells, which might be the cause of drug resistance by DNA repair [16, 17]. There are not enough systematic studies yet compared to HCC; still these results signify USPs as the decisive factors in the development of aggressive traits in CCA. The precise function of USPs in gallbladder cancer is not defined yet, but preliminary evidence suggests USP33 and USP10's involvement in the sustaining of oncogenic signaling [18, 19]. There is a pressing need for further investigations into the USP-directed pathways in GBC due to its infrequent occurrence and the limited number of patient samples.
USPs have been associated not only with their direct actions on tumor cells but also with the aetiology and tumor microenvironment (TME) of HBCs. Among the several USPs, USP7, USP22, USP4, and USP10 are involved in viral replication and chronic inflammatory signaling in the viral hepatitis context; thus, linking infection to cancer development [20, 21]. Moreover, the immunological evasion, the main characteristic of resistance to immunotherapy is supported by USP-mediated stabilization of immune checkpoint regulators. Such results indicate that besides nurturing the tumor's internal development, USPs are also the ones who regulate the external factors that affect the disease's course [22].
Proteases specific to ubiquitin are the new molecular drivers of hepatobiliary malignancies. Their diverse roles in regulating immunological signaling, metabolism, DNA repair, apoptosis, and cell cycle control not only underscore their potential as therapeutic targets but also highlight their varied roles in controlling these processes. Crucially, preclinical research on pharmacological inhibition of USPs is starting to show promise, which could lead to clinical translation. In this review, we summarise the most recent data about USP dysregulation in HCC, CCA, and GBC, investigate their potential as novel therapeutic targets in hepatobiliary malignancies, and look at their molecular roles in oncogenesis and therapeutic resistance.
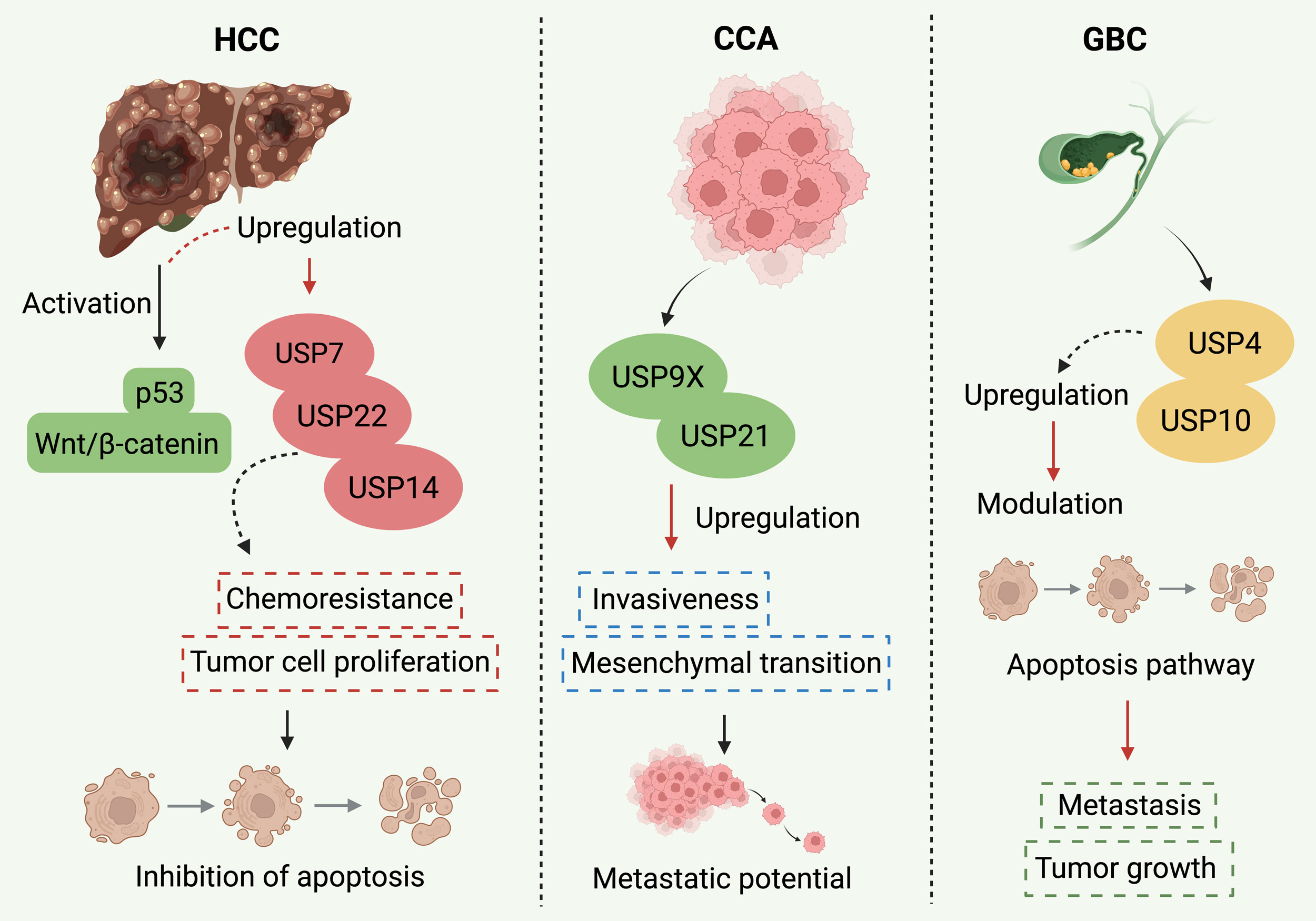 Figure 1. USPs have each taken on different responsibilities in the context of HCC, CCA, and GBC. In the case of HCC, a number of USPs, namely USP7, USP14, and USP22, are being overexpressed and thus they help the cancer cells to grow, become resistant to chemotherapy and finally lead to their death mainly through p53 degradation and Wnt/β-catenin signaling activation. On the other hand, in CCA, USP9X and USP21 are the ones that support the progression to more malignancy by inducing the EMT and thus the cancer cells are more prone to invade. USPs have a different role in GBC, where USP4 and USP10 are also active contributors to the tumor by adopting an apoptosis-related pathway. The depiction of cancer-specific pathways underlies a great variability of USPs in terms of function and that is why the development of specific inhibitors targeting these USPs is proposed in the context of hepatobiliary cancer treatment.
Figure 1. USPs have each taken on different responsibilities in the context of HCC, CCA, and GBC. In the case of HCC, a number of USPs, namely USP7, USP14, and USP22, are being overexpressed and thus they help the cancer cells to grow, become resistant to chemotherapy and finally lead to their death mainly through p53 degradation and Wnt/β-catenin signaling activation. On the other hand, in CCA, USP9X and USP21 are the ones that support the progression to more malignancy by inducing the EMT and thus the cancer cells are more prone to invade. USPs have a different role in GBC, where USP4 and USP10 are also active contributors to the tumor by adopting an apoptosis-related pathway. The depiction of cancer-specific pathways underlies a great variability of USPs in terms of function and that is why the development of specific inhibitors targeting these USPs is proposed in the context of hepatobiliary cancer treatment.
DUBs, which eliminate ubiquitin moieties from proteins or modify polyubiquitin chains, counteract the effects of ubiquitination since it is reversible (Table 1). In addition to protecting proteins against deterioration, these enzymes also recycle ubiquitin molecules and alter the strength or length of signalling cascades. Based on their catalytic domains, the more than 100 deubiquitinases that have been found in humans are divided into several families, such as ovarian tumour proteases (OTUs), USPs, Machado-Joseph disease proteases (MJDs), ubiquitin C-terminal hydrolases (UCHs), JAB1/MPN/MOV34 metalloproteases (JAMMs), MINDY proteases, and ZUFSP family members [26, 27]. Among these, the USP family is the largest and most diverse, comprising approximately 60 members with broad and context-dependent substrate specificity.
The structural defining feature of USPs is a conserved catalytic domain, which is organized in a hand-like architecture with subdomains for palm, thumb, and finger. The structure creates a flexible binding pocket that could potentially recognize diverse substrate proteins and ubiquitin links. The enzymatic activity is facilitated by the cysteine-histidine-aspartate catalytic triad; nevertheless, several USPs are still inactivated by holding conformations until activation by conformational change or cofactor interactions [8, 28]. Various mechanisms involved in the regulation include stress-induced relocalization, interactions with binding partners like the USP1-UAF1 complex, post-translational modifications such as phosphorylation or SUMOylation, and overexpression of the gene in cancer cells. For instance, upon oxidative stress, USP10 is reported to transfer between the cytoplasm and the nucleus where it influences p53 stability [29].
Oncogenesis and the biological roles of USPs are closely related. Many USPs function as tumour promoters by stabilising metabolic enzymes, signalling intermediates, or oncogenic transcription factors. Classic examples include USP22, a histone deubiquitinase that regulates gene expression programs linked to stemness and EMT, and USP7, which suppresses apoptosis by regulating the MDM2-p53 axis [36]. Through their respective modulations of PI3K/AKT and Wnt/β-catenin signalling, which are essential for hepatobiliary carcinogenesis, USP14 and USP9X aid in the advancement of malignancy. However, several USPs have tumor-suppressive effects based on the mutational background and cellular environment. For instance, under genotoxic stress, USP10 can stabilise wild-type p53 and induce apoptosis; yet, when p53 is not functioning or signalling circumstances are changed, it may instead promote oncogenic survival pathways. The intricacy of USP biology and the necessity of carefully assessing its functions in cancer are highlighted by this paradox [37].
Because the liver, biliary system, and gallbladder are constantly exposed to pathogens, xenobiotics, and metabolic byproducts, these are organs that are particularly reliant on proteostasis. Therefore, the UPS and especially USPs play a crucial role in controlling how cells react to damage, infection, and metabolic imbalance in these tissues. The biology of gallbladder cancer is still largely unknown; however, dysregulation of USPs can accelerate CCA progression, cause hepatocarcinogenesis, and additional damage [14]. The abnormal USP expression in HCC regulates apoptosis resistance, angiogenesis, tumour cell proliferation, and treatment resistance. Studies that have been conducted recently point out that the USPs play a role in the metabolic plasticity and DNA repair of CCA, while there is an increasing amount of evidence that USP is involved in the signalling of carcinogenesis in GBC, but comprehensive studies are still lacking. Among the USPs, some are associated with viral hepatitis, which poses a major risk of developing HCC. As an example, USP7 and USP22 have been shown to be related to the persistence and replication of viruses, connecting the viral infection to the process of hepatocarcinogenesis [38, 39].
USPs are the regulators of the ubiquitin system protein stability and signalling integrity, which is a finely tuned system of regulation. Tumour initiation, development, and resistance pathways are largely dependent on the coordination of the ubiquitin system; thus, their dysregulation has dire consequences for cancer biology, particularly in the case of hepatobiliary tumours. Understanding the structural, molecular, and environmental factors that shape the USPs activity is a prerequisite to considering them for therapeutic targeting. In the subsequent sections of this review, the specific USPs dysregulation in GBC, CCA, and HCC will be elaborated further with the implications for clinical translation and their mechanistic significance being emphasized [40].
|
Table 1. Classification of deubiquitinases and representative USPs in HBCs. |
|||||
|
DUB family |
USPs |
Catalytic domain |
Biological functions |
HBC involvement |
References |
|
USP |
USP7, USP10, USP14, USP22, USP9X |
Cysteine protease |
Protein stabilization, signaling regulation, DNA repair, chromatin remodeling |
HCC, CCA, GBC |
[30, 31] |
|
OTU |
OTUD1, OTUD7B |
Cysteine protease |
NF-κB signaling, inflammation |
HCC, CCA |
[32] |
|
MJD |
ATXN3, JOSD1 |
Cysteine protease |
Protein quality control |
HCC |
[33] |
|
UCH |
UCHL1, UCHL3 |
Cysteine protease |
Proteasomal targeting, neuroprotective roles |
HCC |
[34] |
|
JAMM |
BRCC36, Rpn11 |
Metalloprotease |
DNA repair, proteasome regulation |
HCC, CCA |
[35] |
By stabilising carcinogenic proteins or altering survival pathways, several USPs function as molecular drivers of carcinogenesis in HCC (Figure 2). For example, USP9X, which is often overexpressed in HCC, protects β-catenin from proteasomal degradation, enhancing Wnt/β-catenin signalling. This promotes proliferation and is associated with a lower patient survival rate [47]. Similarly, USP7 has been shown to stabilise MDM2, which results in p53 degradation and reduced apoptotic responses. This mechanism gives hepatocytes resilience to treatments that damage DNA [48]. It has been demonstrated that USP22, a member of the SAGA complex, deubiquitinates histones H2A and H2B in HCC, maintaining chromatin states that support stemness-like phenotypes, EMT, and oncogenic transcriptional programs [49, 50]. USP22 is a possible prognostic biomarker since clinical evaluations consistently show that increased expression of the protein in HCC tissues correlates with advanced stage, vascular invasion, and poor overall survival (Figure 3, 4).
USP10 is another important USP in HCC. Its function exemplifies the dual nature of USPs in cancer biology: although it can occasionally stabilise and activate wild-type p53, research on HCC has shown that USP10 may also shield oncogenic substrates like YAP/TAZ, promoting tumour growth and invasion [51]. Because it controls the stability of proteins involved in metabolic signalling and cell proliferation, USP14 is equally important. Increased glycolytic flux and hyperactivation of the AKT/mTOR pathway have been linked to elevated USP14 expression in HCC, highlighting the connection between deubiquitination and liver metabolic rewiring [52].
In addition to these extensively researched instances, several other USPs have been linked to hepatocarcinogenesis. It has been demonstrated that USP5 controls NF-κB signalling, which promotes the growth of tumours driven by chronic inflammation [53, 54]. Under metabolic stress conditions typical of cirrhotic livers, USP13 provides growth advantages by stabilising mitochondrial proteins that promote oxidative phosphorylation [55, 56]. According to reports, USP19 enhances angiogenesis and adaptability to the hypoxic tumour microenvironment via controlling hypoxia-inducible factor 1-alpha (HIF-1α) [57, 58]. All of these studies show that the dysregulation of several USPs, each of which targets different substrates but converges on the characteristics of malignancy, characterises the HCC landscape.
A less well-studied condition, cholangiocarcinoma, has also been connected to abnormal USP activity. One such example is USP21, which improves metabolic adaptability in CCA cells and accelerates tumour growth by stabilising glycolytic enzymes like ENO1 [59]. By deubiquitinating PARP1, USP1 contributes equally to the prolonged DNA repair activity that allows tumour cells to resist the genotoxic stress caused by chemotherapy [16]. By controlling Smad4, USP9X has also been linked to CCA, indicating a part in invasion and metastasis triggered by TGF-β [60]. According to recent research, USP39, an RNA splicing factor-associated DUB, contributes to the aberrant cell-cycle progression in CCA. In CCA cell lines, USP39 knockdown has been demonstrated to suppress proliferation and trigger apoptosis [34, 61]. Together, our results imply that USPs support metabolic and epigenetic modifications that maintain the progression of cancer in addition to intrinsic tumour cell survival in CCA.
Although there is still little research on GBC in relation to deubiquitination, new findings suggest that some USPs play significant roles. According to research, USP33 modulates receptor tyrosine kinase signalling, and its absence causes GBC cells' MAPK pathways to become hyperactivated [62]. In gallbladder cancer, USP10 has also been shown to maintain oncogenic NF-κB signalling, which connects inflammation to the growth of the tumour [63]. These early findings highlight the possibility that GBC, like HCC and CCA, is impacted by abnormal deubiquitination processes that may one day be used for therapeutic benefit, even though the research is still preliminary [64].
It is crucial to understand that USP dysregulation in hepatobiliary malignancies affects the tumour microenvironment and resistance to treatment in addition to intrinsic oncogenic signalling. For instance, USP7 has been linked to immune evasion, PD-L1 stabilisation, and decreased immune checkpoint blockade effectiveness in HCC [65]. Despite being an ISG15-specific protease, USP18 functions similarly to USPs and has been demonstrated to control interferon signalling in HCC, which helps to inhibit the immune system [66]. According to Zhang et al. (2023), USP21-mediated stabilisation of inflammatory mediators in CCA supports a tumor-permissive stroma by facilitating interaction with cancer-associated fibroblasts. These observations highlight the broader influence of USPs in shaping not just cancer cells themselves but also the ecosystem in which they thrive [60].
In the case of HBCs, USP dysregulation has been linked to aggressive disease and resistance to treatment from the clinical perspective. For example, in HCC, poor response to sorafenib was correlated with elevated USP14 expression, while USP22 expression was linked to shorter overall survival after surgical resection [67, 68]. On the other hand, USP1 expression was found to be associated with resistance to platinum-based chemotherapy in CCA [69]. These findings are indicative of the potential role of USPs as biomarkers for prediction and they will possibly guide clinicians in the selection of treatments along with their potential use as molecular drivers.
The data have given strong support to the idea that dysregulated ubiquitin-specific proteases are the main molecular drivers in GBC, CCA, and HCC. Their participation in the development of cancers in the liver and biliary tract is evidenced by their ability to manage oncogenic signalling, metabolism, DNA repair, immunological evasion, and treatment resistance. The diversity of their functions also allows for a supposition that concentrating on certain USPs or their substrates might provide new routes for therapeutic intervention; this idea is further elaborated in the following sections.
|
Table 2. Dysregulated USPs and their mechanistic roles in HBCs. |
|||||
|
USP |
Cancer type |
Pathway |
Mechanistic role |
Clinical correlation |
References |
|
USP21 |
CCA |
ENO1, glycolytic enzymes |
Metabolic adaptation |
Tumor progression |
[15] |
|
USP7 |
HCC |
MDM2-p53, PD-L1 |
Immune evasion, apoptosis suppression |
Poor survival, therapy resistance |
[34] |
|
USP10 |
HCC, GBC |
p53, YAP/TAZ |
Cell survival, invasion |
Advanced stage, metastasis |
[42] |
|
USP14 |
HCC |
AKT/mTOR |
Metabolic reprogramming, proliferation |
Sorafenib resistance |
[43] |
|
USP22 |
HCC |
H2A/H2B, EMT genes |
Chromatin remodeling, stemness |
Aggressive tumors, poor prognosis |
[44] |
|
USP1 |
CCA |
PARP1, FANCD2 |
DNA repair, chemoresistance |
Platinum resistance |
[45] |
|
USP9X |
HCC, CCA |
β-catenin, Smad4 |
Wnt signaling, TGF-β signaling |
Poor survival, invasion |
[46] |
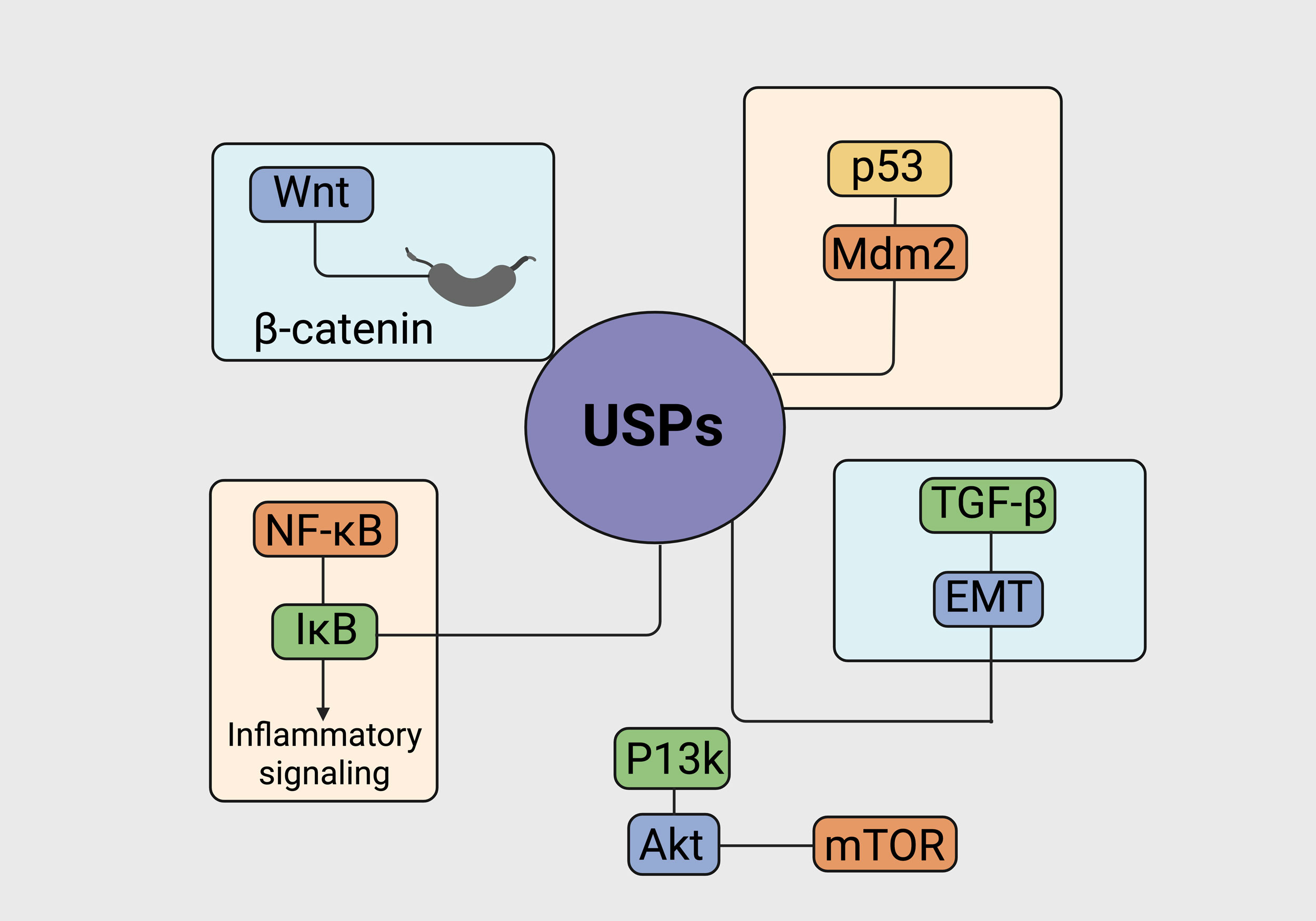 Figure 2. Crosstalk between USPs and important cancer-causing signaling pathways in liver and bile duct malignancies. The sketch depicts the juncture of the USPs that are out of control and the corresponding critical molecular pathways that tumor initiation and progression. The main USPs are involved with and deubiquitinate their key substrates in different signaling areas, thus affecting many cancer-making processes. In the Wnt/β-catenin pathway, USPs are the ones that keep β-catenin in the nucleus because they do not allow it to go through the whole aerobic degradation process by ubiquitin, so the transcription of oncogenic genes is targeted more than usual. In the case of p53/Mdm2, there are some USPs that through deubiquitination of p53, allow for tumor suppression and other USPs which in turn stabilize Mdm2 leading to p53 inactivation and uncontrolled tumor growth. The USPs cap IκB degradation and this way they control NF-κB activation; thus, they are the ones who promote inflammatory signaling and cancer cell survival. USPs are the ones who make the decision whether the degradation of key intermediates in the PI3K/Akt/mTOR pathway will be stopped or not and, thus, play a significant role in the triggering of the downstream signaling cascades that are supportive of the cells being alive and multiplying. Additionally, USPs contribute to EMT in the TGF-β signaling pathway by modulating receptor availability and downstream effectors, ultimately driving invasion and metastatic potential in hepatobiliary malignancies.
Figure 2. Crosstalk between USPs and important cancer-causing signaling pathways in liver and bile duct malignancies. The sketch depicts the juncture of the USPs that are out of control and the corresponding critical molecular pathways that tumor initiation and progression. The main USPs are involved with and deubiquitinate their key substrates in different signaling areas, thus affecting many cancer-making processes. In the Wnt/β-catenin pathway, USPs are the ones that keep β-catenin in the nucleus because they do not allow it to go through the whole aerobic degradation process by ubiquitin, so the transcription of oncogenic genes is targeted more than usual. In the case of p53/Mdm2, there are some USPs that through deubiquitination of p53, allow for tumor suppression and other USPs which in turn stabilize Mdm2 leading to p53 inactivation and uncontrolled tumor growth. The USPs cap IκB degradation and this way they control NF-κB activation; thus, they are the ones who promote inflammatory signaling and cancer cell survival. USPs are the ones who make the decision whether the degradation of key intermediates in the PI3K/Akt/mTOR pathway will be stopped or not and, thus, play a significant role in the triggering of the downstream signaling cascades that are supportive of the cells being alive and multiplying. Additionally, USPs contribute to EMT in the TGF-β signaling pathway by modulating receptor availability and downstream effectors, ultimately driving invasion and metastatic potential in hepatobiliary malignancies.
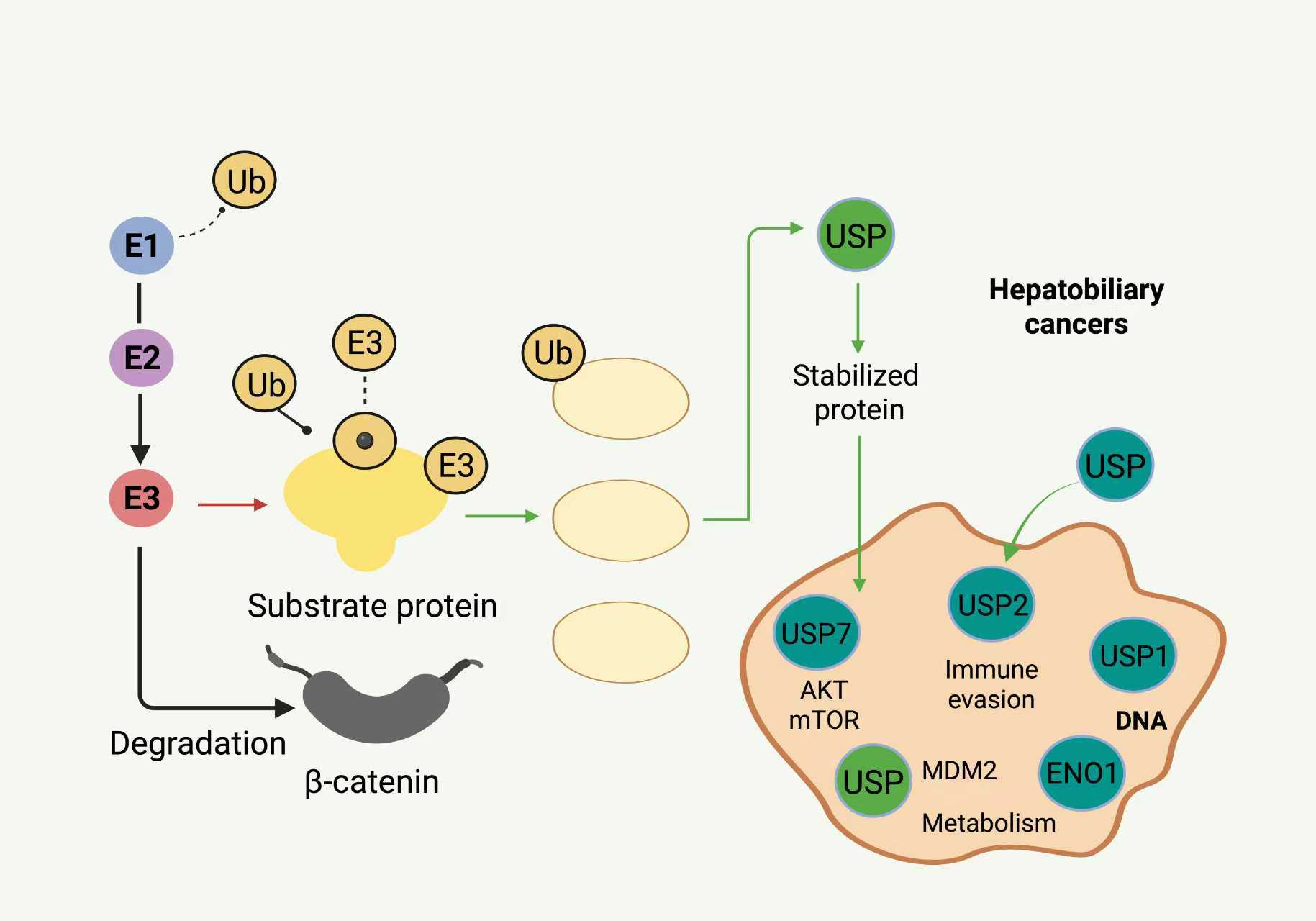 Figure 3. Overview of the UPS and the role of USPs in HBCs. The figure illustrates the process of protein ubiquitination mediated by E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, leading to substrate proteins being targeted for proteasomal degradation. USPs (USP7, USP10, USP14, USP22, USP21, USP1) remove ubiquitin chains from specific oncogenic or tumor suppressor substrates, including β-catenin, MDM2, PD-L1, H2A/H2B, FANCD2, and ENO1. USP-mediated stabilization enables activation of oncogenic signaling (Wnt/β-catenin, AKT/mTOR), DNA repair, metabolic adaptation, chromatin remodeling, and immune evasion. Red arrows indicate ubiquitin-mediated degradation, and green arrows indicate USP-mediated deubiquitination and protein stabilization.
Figure 3. Overview of the UPS and the role of USPs in HBCs. The figure illustrates the process of protein ubiquitination mediated by E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, leading to substrate proteins being targeted for proteasomal degradation. USPs (USP7, USP10, USP14, USP22, USP21, USP1) remove ubiquitin chains from specific oncogenic or tumor suppressor substrates, including β-catenin, MDM2, PD-L1, H2A/H2B, FANCD2, and ENO1. USP-mediated stabilization enables activation of oncogenic signaling (Wnt/β-catenin, AKT/mTOR), DNA repair, metabolic adaptation, chromatin remodeling, and immune evasion. Red arrows indicate ubiquitin-mediated degradation, and green arrows indicate USP-mediated deubiquitination and protein stabilization.
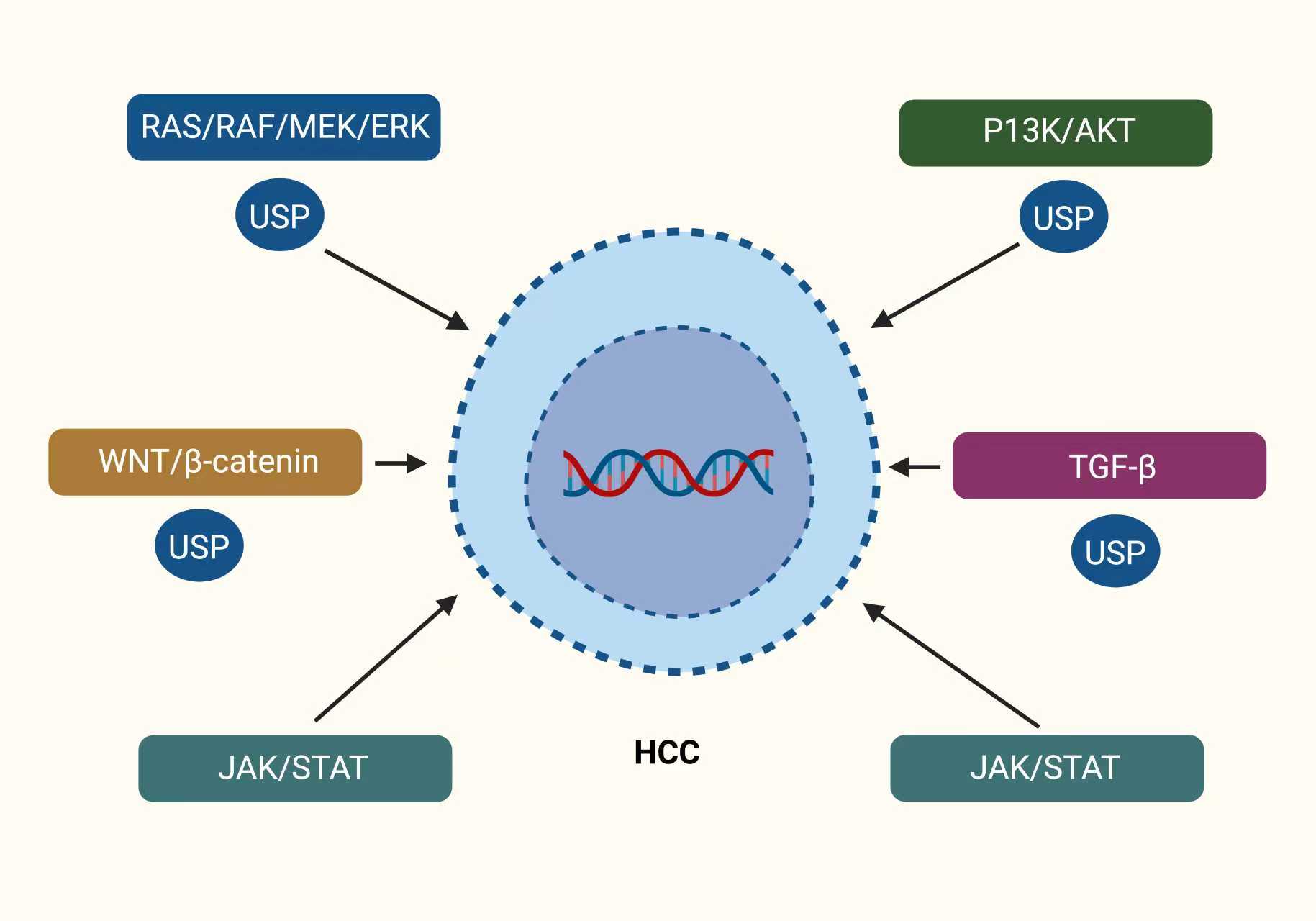 Figure 4. Key oncogenic signaling pathways regulated by the USP family members in HCC. The sketch shows that the USPs are the main players in the regulation of the principal oncogenic pathways in HCC. An HCC cell is portrayed in the middle of the diagram with a nucleus containing DNA, a sign of genetic control. The most important signaling pathways related to cancer, RAS/RAF/MEK/ERK, PI3K/AKT, WNT/β-catenin, JAK/STAT, and TGF-β, are drawn around the cell. The pathways are all linked to the cell by arrows indicating their role in the process of tumor growth, invasion, survival, and metastasis. The oval components marked “USP” point out the different USPs involved in the process of stabilizing with the help of preventing degradation through proteasomal action, the key signaling proteins in each pathway. The illustration highlights that the impaired USPs' deubiquitination process strengthens the oncogenic signaling, thus revealing the possible drug targets for HCC treatment.
Figure 4. Key oncogenic signaling pathways regulated by the USP family members in HCC. The sketch shows that the USPs are the main players in the regulation of the principal oncogenic pathways in HCC. An HCC cell is portrayed in the middle of the diagram with a nucleus containing DNA, a sign of genetic control. The most important signaling pathways related to cancer, RAS/RAF/MEK/ERK, PI3K/AKT, WNT/β-catenin, JAK/STAT, and TGF-β, are drawn around the cell. The pathways are all linked to the cell by arrows indicating their role in the process of tumor growth, invasion, survival, and metastasis. The oval components marked “USP” point out the different USPs involved in the process of stabilizing with the help of preventing degradation through proteasomal action, the key signaling proteins in each pathway. The illustration highlights that the impaired USPs' deubiquitination process strengthens the oncogenic signaling, thus revealing the possible drug targets for HCC treatment.
Stabilisation of oncogenic signalling intermediates is one of the main ways in whereby USPs aid in the advancement of hepatobiliary carcinoma. USP9X is involved in the stabilization of β-catenin in liver cancer cells by obstructing the latter's ubiquitin-mediated degradation and by Wnt pathway activation which in turn opens up new cell divisions and maintains cancer-like characteristics [71]. USP22's removal of ubiquitin from histones H2A and H2B is a similar process of chromatin remodelling, where transcriptionally permissive chromatin that promotes the expression of genes linked to angiogenesis and EMT is maintained [49]. USP14 is said to keep the oncogenic PI3K/AKT signaling on by newly stabilizing the associated upstream kinases and impeding their degradation, hence enhancing cell proliferation and survival [72]. These USPs act together to hold the stability of core pathways that would otherwise be tightly dependent on ubiquitin for their degradation.
USPs exert an equally significant influence on DNA repair and genomic integrity, thus allowing tumor cells to cope with genotoxic stress coming from both therapeutic approaches and internal metabolic byproducts. USP1, for instance, has been a major player in this context by deubiquitinating proteins associated with the Fanconi anaemia pathway, especially FANCD2. This not only maintains DNA crosslink repair but also protects tumorigenic cells from cisplatin-based chemotherapy in CCA [78]. In the case of HCC, USP3 has been associated with the de-ubiquitination of H2AX, thus ensuring fast double-strand break repair and contributing to increased resistance to radiation [79]. These USPs help tumour cells survive genomic instability and also contribute to therapeutic resistance, which is a significant problem in hepatobiliary oncology, by enhancing DNA repair capability.
Another characteristic of cancer progression that is closely related to USP activity is metabolic reprogramming. USP21 has been shown to deubiquitinate ENO1 in cholangiocarcinoma, promoting glycolysis and facilitating quick ATP synthesis even in the presence of nutritional shortages [15]. USP29 promotes metabolic flexibility in HCC by stabilising c-Myc, a crucial transcriptional regulator of glycolytic and glutaminolytic enzymes [80]. By stabilising mitochondrial proteins that sustain oxidative phosphorylation, USP13 aids in metabolic adaptability, which is especially beneficial in microenvironments that are low in oxygen and nutrients [43]. These results show that USPs alter the metabolic landscape of tumours and control signalling proteins, allowing HBC cells to proliferate in unfavourable microenvironments.
The control of cell death pathways is another major theme. In HCC, for example, USP10 stabilises mutant p53, changing its role from tumour suppression to a gain-of-function oncogene that encourages invasion and chemoresistance [37]. Although less research has been done on USP2 in hepatobiliary tumours, it has been demonstrated to stabilise MDM2, which lowers p53 levels and prevents HCC cells from undergoing apoptosis [34]. Moreover, USP5, by the removal of ubiquitin chains from IκBα, boosts NF-κB activation, which, in turn, limits the degradation of NF-κB and allows for the sustained pro-survival inflammatory signalling that continues to be attractive in the case of cancer [81]. If we consider these mechanisms all together, they provide a way for tumour cells to dodge apoptotic checkpoints and keep inflammation going, which are the two main processes that promote cancer growth.
USPs act as mediators of stromal and immunological interactions and the TME is gradually recognized as having a decisive role in the whole scenario of hepatobiliary tumors. A major role in immune evasion and in diminishing the effectiveness of immune checkpoint inhibitors is attributed to USP7 through the stabilization of PD-L1 on HCC cells [82]. Likewise, the USP18 helps the tumor cells avoid death via immune response by deubiquitinating the necessary signaling intermediates that can through energy consumption by the tumor cell inhibit the immune response [83]. The extracellular matrix remodelling and fibroblast activation in CCA are associated with USP21, which creates an environment that supports tumor growth and infiltration [84]. These findings indicate that USPs not only keep tumor-intrinsic oncogenic pathways active but also, in fact, take an active part in influencing the immunosuppressive and desmoplastic microenvironments of the tumors. By stabilizing PD-L1 on the surface of HCC cells, USP7 has a particularly significant function in facilitating immune evasion and decreasing the efficacy of immune checkpoint inhibitors [85]. Similarly, by deubiquitinating important signaling intermediates, USP18 inhibits interferon responses, allowing tumor cells to avoid immune-mediated death [86].
Through extracellular matrix breakdown, cytoskeletal remodelling, and EMT, USPs contribute to metastasis. By controlling transcriptional pathways linked to vimentin induction and E-cadherin suppression, USP22 promotes EMT [87]. By stabilising proteins involved in actin filament remodelling, USP10 promotes invasion and dissemination and increases cytoskeletal flexibility [88]. Furthermore, it has been demonstrated that USP39, which was previously linked to the advancement of the CCA cell cycle, controls splice variants of genes linked to cell motility, establishing a connection between RNA processing and the capacity for metastasis [89]. These investigations all share the finding that USP activity converges on biological mechanisms that enable HBC cells to infiltrate, colonise, and detach from distant organs.
One of the most therapeutically significant effects of USP activity is drug resistance. Resistance to sorafenib, the most popular systemic treatment for advanced HCC has been closely linked to USP7 and USP14. Mechanistically, they lessen drug-induced apoptosis by stabilising pro-survival pathways [90]. Resistance to metabolic inhibitors in HCC has been associated with USP29-mediated stabilisation of c-Myc [34]. These findings demonstrate how USPs mediate adaptive resistance, and they imply that pharmacologically targeting USPs may enhance current treatments and get around present clinical management constraints.
A variety of molecular processes regulated by ubiquitin-specific proteases coordinate the development of hepatobiliary malignancies. USPs serve as the molecular builders of malignancy,
influencing everything from metabolic rewiring, apoptosis evasion, microenvironment manipulation, and treatment resistance to oncogenic signalling stabilisation and DNA repair reinforcement. They have an impact on tumour ecosystems and cellular compartments, which emphasises the necessity of treating them as both potential treatment targets and biomarkers of progression. Transforming USP biology into real clinical advantages for patients with hepatobiliary malignancies will require a more thorough analysis of these mechanistic functions as research progresses [91].
|
Table 3. Summary of USP-mediated mechanisms driving HBCs progression. |
|||||
|
Mechanism |
USPs |
Downstream effects |
Cancer type |
Implications |
References |
|
Metabolic reprogramming |
USP21, USP29, USP13 |
Glycolysis, oxidative phosphorylation |
HCC, CCA |
Target metabolic vulnerabilities |
[34] |
|
Microenvironment modulation |
USP21 |
Fibroblast activation, ECM remodeling |
CCA |
Disrupt stromal support |
[71] |
|
Oncogenic signaling |
USP9X, USP22, USP14 |
Wnt/β-catenin, AKT/mTOR, EMT |
HCC, CCA |
Combination with pathway inhibitors |
[73] |
|
DNA repair |
USP1, USP3 |
FANCD2, H2AX stabilization |
HCC, CCA |
Sensitize to chemotherapy/radiotherapy |
[74] |
|
Apoptosis evasion |
USP10, USP2 |
p53 stabilization/inactivation |
HCC, GBC |
Restore apoptosis with USP inhibitors |
[75] |
|
Immune evasion |
USP7, USP22, USP18 |
PD-L1, interferon signaling |
HCC |
Combine with immunotherapy |
[76] |
|
Metastasis |
USP22, USP10, USP39 |
EMT, cytoskeletal remodeling, splicing |
HCC, CCA |
Reduce invasion/metastasis |
[77] |
The most sophisticated USP-targeting substances in preclinical research are USP7, USP14, and USP1 inhibitors. In HCC, where USP7 maintains immune evasion by stabilising PD-L1 and suppressing antitumor immunity, USP7 inhibitors show great promise. The USP7–MDM2–p53 axis can be disrupted by novel small compounds like FT671 and its analogues, which reactivates apoptotic signalling [93]. Pharmacologic USP7 inhibition has demonstrated therapeutic significance in HCC xenograft models by causing tumour regression and increased susceptibility to immune checkpoint inhibitors [30]. In preclinical models of HCC, USP14 inhibitors like IU1 and b-AP15 have also demonstrated effectiveness by decreasing proteasome-associated deubiquitination, which causes oncogenic substrates to degrade and sorafenib sensitisation [31]. Notably, adaptive resistance mechanisms that restrict the persistence of tyrosine kinase inhibitor responses seem to be circumvented by USP14 inhibition.
The attention towards USP1 inhibitors in CCA is due to USP1, the protease that maintains PARP1 and FANCD2, which are the two pathways through which DNA can be repaired, thus making cells susceptible to the actions of the inhibitors. When a very small molecule, such as ML323, disrupts the USP1-UAF1 interaction, CCA cells become more susceptible to platinum therapy and PARP inhibitors [96]. The conception of DNA damage repair spread is now being validated with the co-utilization of USP1 inhibitors and genotoxic treatments in preclinical studies, as the latter has been notorious for their unregulated repair mechanisms in CCA. USP22, a deubiquitinating enzyme associated with chromatin, is a TOC candidate too; it is connected to EMT, stemness, and drug resistance. Even though RNA interference and CRISPR-based experimentation have shown that USP22 knockdown leads to reduced aggressiveness and resensitization of HCC cells to sorafenib and lenvatinib, the selective USP22 blockers are still in their infancy [98].
In addition to direct inhibitors, PROTACs which are intended to break down USPs are gaining popularity. In contrast to catalytic inhibitors, PROTACs that target USP7 have been successfully designed to promote ubiquitination and proteasomal clearance of USP7 itself, resulting in greater anticancer efficacy [99]. With the benefit of removing scaffolding activities that persist after enzymatic inhibition, such strategies may potentially be applied to additional oncogenic USPs. Likewise, covalent inhibitors that target USPs like USP10 and USP9X's catalytic cysteine residues are being studied; preliminary compounds have demonstrated selective efficacy in HCC models [95].
Modulating the indirect effects of USP activity is another aspect of therapeutic approaches. For example, USP9X inhibition can work in concert with inhibitors of the Wnt/β-catenin pathway since USP9X stabilises β-catenin in HCC. USP9X inhibitors have demonstrated more potent anticancer effects in preclinical animals when combined with either tankyrase inhibitors or porcupine inhibitors than when used alone [71]. Similarly, USP-driven glycolysis in CCA creates metabolic vulnerabilities that can be exploited by combining USP21 targeting with glycolytic inhibitors, which reduces tumour growth in xenografts [15]. These examples show how sensible combinatorial regimen design is made possible by a grasp of USP-mediated pathways.
Since USPs also control vital biological functions in healthy tissues, the possible toxicity of USP inhibition is a crucial factor to take into account when translating therapeutics. Developing inhibitors with improved selectivity for USP conformations particular to cancer or taking advantage of tumor-specific cofactors that interact with USPs are two ways to deal with this. For instance, inhibitors that target this relationship seem to exhibit preferential activity in cancer cells with severe replication stress, as USP1 requires UAF1 for complete activity [100]. Another strategy is the creation of context-dependent PROTACs, which reduce off-target toxicity by selectively degrading USPs in tumour cells that express particular E3 [101].
Another area where USP-targeting could have a game-changing effect is immunotherapy. Poor responses to immune checkpoint blockage in HCC can be explained mechanistically by the stabilisation of PD-L1 mediated by USP7 and USP22. According to preclinical research, cytotoxic T-cell infiltration and tumour clearance are improved when USP inhibitors are used with anti-PD-1 or anti-PD-L1 antibodies [102]. Furthermore, USP18's function in inhibiting interferon signalling suggests that innate immunity against hepatobiliary tumours may be strengthened by its suppression. Therefore, therapeutic targeting of USPs may address one of the most important issues in these cancers and supplement current immunotherapies.
There are currently no licensed drugs specifically treating hepatobiliary malignancies, and clinical translation of USP inhibitors is still in its early stages. Nonetheless, a number of first-in-human studies involving USP inhibitors in solid tumours and haematologic malignancies are currently in progress, offering important pharmacokinetic and safety information. Future research in HCC, CCA, and GBC will be guided by the lessons learnt from these early trials, especially in the areas of dose-limiting toxicities and biomarker identification [103]. Because USP expression levels or activity profiles may identify subsets of patients most likely to benefit, biomarker-driven patient stratification will be crucial. For instance, USP7 or USP22 expression may be prognostic biomarkers for how well HCC responds to USP inhibitors. Similarly, USP1-targeted treatments in conjunction with PARP inhibitors or platinum drugs may be guided by genomic profiling of DNA damage repair defects in CCA [104].
There is considerable potential for incorporating USP-targeted tactics into multimodal treatment approaches. While innovative drug delivery methods, such as nanoparticle carriers may lessen systemic toxicity, USP inhibitors may be able to overcome innate and acquired resistance when combined with kinase inhibitors, immunotherapy, or metabolic treatments. The range of selective USP inhibitors that are available for preclinical and clinical testing will also continue to grow as a result of developments in structural biology and computational drug design. Ultimately, a careful balance between safety and efficacy, bolstered by mechanistic discoveries and biomarker-driven clinical trials, will be necessary to translate USP biology into therapeutic benefit for patients with hepatobiliary malignancies [105].
|
Table 4. Current USP-targeting therapeutic approaches in HBCs. |
|||||
|
USP |
Inhibitor |
Mechanism |
Model |
Approaches |
References |
|
USP9X |
Covalent inhibitors |
Destabilize β-catenin |
HCC |
Wnt/β-catenin inhibitors |
[71] |
|
USP7 |
FT671, PROTACs |
Inhibit MDM2 stabilization, degrade USP7 |
HCC xenografts |
Anti-PD-1/PD-L1 immunotherapy |
[94] |
|
USP14 |
IU1, b-AP15 |
Inhibit proteasome-associated DUB activity |
HCC |
Sorafenib or lenvatinib |
[95] |
|
USP1 |
ML323 |
Disrupt USP1-UAF1 complex, inhibit DNA repair |
CCA |
Cisplatin, PARP inhibitors |
[96] |
|
USP22 |
RNAi, CRISPR |
Knockdown of chromatin-associated activity |
HCC |
Sorafenib, metabolic inhibitors |
[97] |
 Figure 5. Therapeutic targeting of USPs in HBCs. Upper Left: selective small-molecule inhibitors (FT671, IU1, ML323) and PROTACs target specific USPs in tumor cells within the liver. Center: inhibition or degradation of USPs disrupts oncogenic ubiquitin signaling, leading to increased tumor-cell death and enhanced vulnerability to co-treatments. Right: Combinatorial strategies immune checkpoint blockade (immunotherapy), standard cytotoxic agents (chemotherapy), and targeted kinase inhibitors are proposed to cooperate with USP inhibition, resulting in tumor regression and therapy sensitization. Icons indicate representative clinical outcomes.
Figure 5. Therapeutic targeting of USPs in HBCs. Upper Left: selective small-molecule inhibitors (FT671, IU1, ML323) and PROTACs target specific USPs in tumor cells within the liver. Center: inhibition or degradation of USPs disrupts oncogenic ubiquitin signaling, leading to increased tumor-cell death and enhanced vulnerability to co-treatments. Right: Combinatorial strategies immune checkpoint blockade (immunotherapy), standard cytotoxic agents (chemotherapy), and targeted kinase inhibitors are proposed to cooperate with USP inhibition, resulting in tumor regression and therapy sensitization. Icons indicate representative clinical outcomes.
The context-dependent duality of the USP function is another significant obstacle. A number of USPs, such as USP10 and USP22, can either promote or prevent tumour growth, depending on the type of tissue, cellular stressor, or mutational landscape. While USP10 facilitates DNA damage repair and p53-dependent apoptosis in normal hepatocytes, it may stabilise mutant p53 in hepatocellular carcinoma, encouraging invasion and drug resistance [63]. In order to prevent unforeseen repercussions from USP-targeted therapies, this duality generates a fragile therapeutic window that necessitates careful patient selection and mechanistic understanding. Predicting clinical outcomes is made more difficult by the fact that preclinical models frequently fall short of accurately simulating the variety of real hepatobiliary tumours.
The less-studied hepatobiliary malignancies, especially GBC, have a significant knowledge gap. Although new research links USPs like USP10 and USP33 to the development of GBC, there is still a lack of thorough profiling of USP expression, substrate networks, and mechanistic contributions [106]. The systematic mapping of the USP-substrate landscape is also lacking, despite the fact that CCA research has identified USPs such as USP1, USP21, and USP39 as drivers of DNA repair, metabolism, and splicing. To close these gaps and identify the entire repertoire of USPs active in each hepatobiliary malignancy, integrative techniques integrating transcriptomics, proteomics, and functional genomics will be necessary.
Rapid clinical translation is further hampered by pharmacologic issues. Although USP7, USP14, USP1, and USP22-targeting selective inhibitors and PROTACs have demonstrated encouraging preclinical efficacy, questions still surround their safety profile, pharmacokinetics, and tumor-specific delivery. In non-tumor tissues, USPs control vital biological functions, increasing the risk of off-target harm. Through the development of tumour-specific delivery methods such as context-dependent PROTACs, antibody–drug conjugates, or nanoparticles, systemic adverse effects could be reduced significantly. Furthermore, it might be necessary to apply biomarker-guided patient selection and adaptive dosage techniques because of the dynamic changes in USP activity throughout tumour progression or medication exposure.
The yet not so serious issues do not reduce the number of highly interesting ways for research that come next. To start with, a great mapping of USP-substrate interactions in HBCs along with the application of the latest ubiquitinomics and proteomics technologies may open up new avenues for therapeutics. Moreover, the use of USP inhibition alongside existing treatments is going to be a major focus of combination strategies. To combat inherent or acquired resistance and improve clinical outcomes, for example, USP1 or USP7 inhibitors could be combined with metabolic inhibitors, DNA-damaging agents, or immunotherapy [107]. Also, the development of predictive biomarkers based on cofactor dependencies, activity signatures, or USP expression may pave the way for precision medicine. This would ensure that only those patients with the highest likelihood of benefiting from USP-targeted therapies would be treated with them.
Another fascinating way is through the exploitation of the TME. The targeting of USPs could possibly augment the efficacy of immunotherapy or interrupt tumor-supportive areas as they modify inflammatory signaling, stromal interactions, and immune checkpoint mechanisms. A case in point, in hepatocellular carcinoma, USP7 and USP22 act to reduce the function of cytotoxic T-cells and at the same time stabilize PD-L1, providing a mechanistic rationale for the use of USP inhibitors together with checkpoint blockade [108]. Targeting USP21-mediated fibroblast activation in CCA may also improve chemotherapeutic penetration and lessen desmoplasia [109]. Gaining insight into these microenvironmental functions may increase USP inhibitors' usefulness beyond their ability to directly target tumour cells.
Lastly, achieving the therapeutic potential of USPs will require technological advancements in medication delivery and design. Covalent inhibitors, PROTAC-based degradation techniques, and structure-guided drug discovery are all developing quickly and present chances to target tumor-relevant USPs specifically while reducing toxicity. Finding highly selective compounds may be sped up by combining machine learning and artificial intelligence to optimise the structure–activity connection. Simultaneously, the creation of reliable organoids, humanised mouse systems, and patient-derived models will allow preclinical assessment of safety, efficacy, and combinatorial regimens in settings more representative of HBCs in humans.
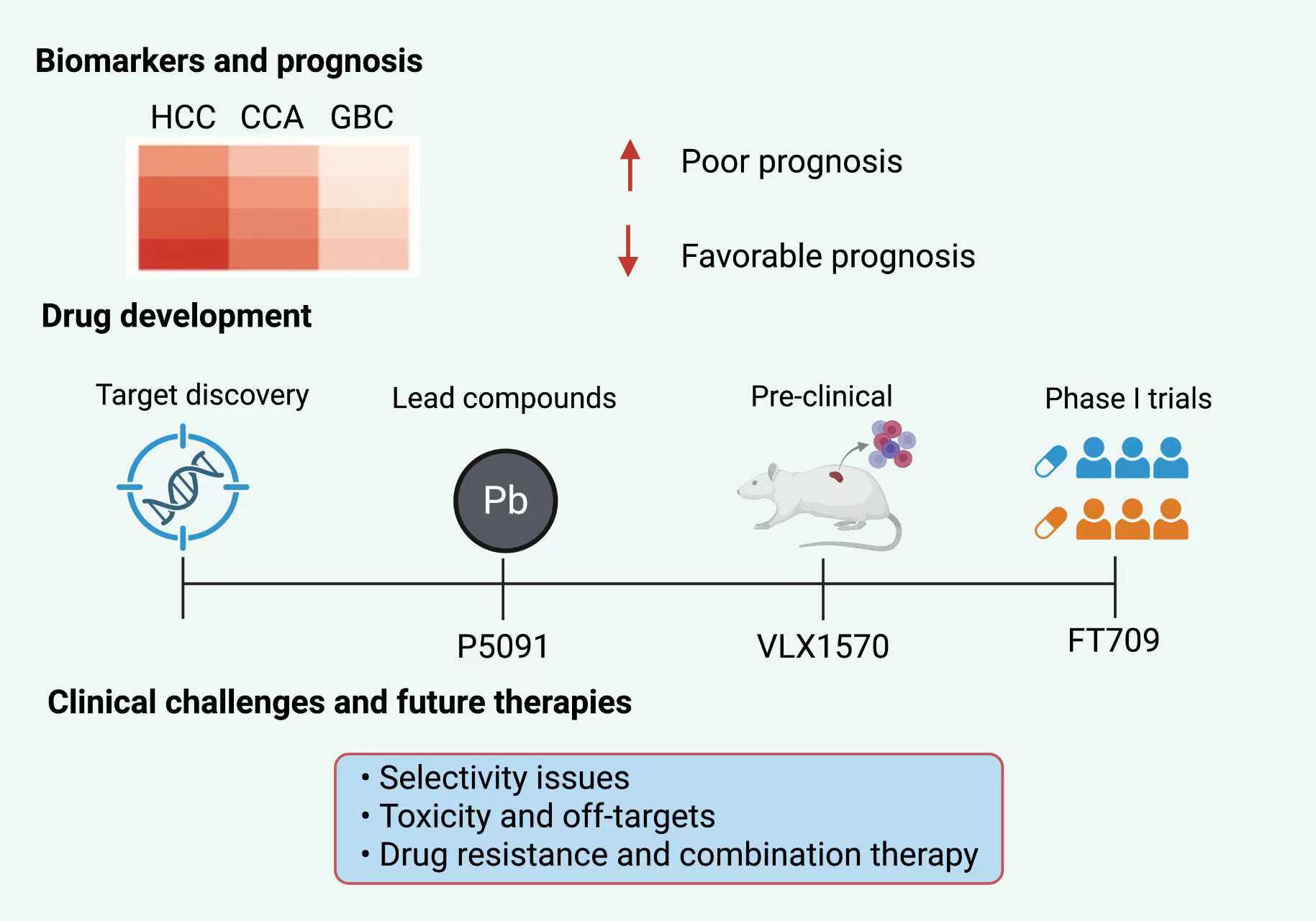 Figure 6. The clinical and translational landscape of USPs in HBCs is depicted in this figure. This figure highlights the growing clinical importance of USPs in HCC, CCA, and GBC. The top panel showcases a heatmap that shows different levels of USP expression in various types of liver and bile duct tumors. The illustration indicates that the presence of higher USP levels correlates with poor survival outcomes, while lower expression may be a sign of a favorable prognosis. The middle panel presents a timeline for the development of drugs which is dedicated to the current progress made by USP-targeted therapeutics, including lead compounds like P5091 (USP7 inhibitor), VLX1570 (USP14/USP30 inhibitor), and FT709 (USP16/USP28 inhibitor). Their journey through different phases of research is shown going from the discovery of the target to preclinical studies and then early-phase trials. The bottom panel discusses the major clinical challenges which are defined as selectivity of inhibitors, toxicity to non-targets, and the development of drug resistance, thus pointing out the necessity for combination strategies and the application of precision medicine approaches in the future USP-based therapy scenario.
Figure 6. The clinical and translational landscape of USPs in HBCs is depicted in this figure. This figure highlights the growing clinical importance of USPs in HCC, CCA, and GBC. The top panel showcases a heatmap that shows different levels of USP expression in various types of liver and bile duct tumors. The illustration indicates that the presence of higher USP levels correlates with poor survival outcomes, while lower expression may be a sign of a favorable prognosis. The middle panel presents a timeline for the development of drugs which is dedicated to the current progress made by USP-targeted therapeutics, including lead compounds like P5091 (USP7 inhibitor), VLX1570 (USP14/USP30 inhibitor), and FT709 (USP16/USP28 inhibitor). Their journey through different phases of research is shown going from the discovery of the target to preclinical studies and then early-phase trials. The bottom panel discusses the major clinical challenges which are defined as selectivity of inhibitors, toxicity to non-targets, and the development of drug resistance, thus pointing out the necessity for combination strategies and the application of precision medicine approaches in the future USP-based therapy scenario.
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
Enas Roumieh conceived the topic and outline of the review. Waqas Bin Ismail performed the literature search, data collection, and critical analysis of the included studies. Samahir Sheikh Idris provided conceptual guidance, supervised the overall writing process, and revised the manuscript for important intellectual content. All authors read and approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018, 68(6): 394-424.
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F: Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021, 71(3): 209-249.
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A: Global cancer statistics, 2012. CA Cancer J Clin 2015, 65(2): 87-108.
- Lin L, Li Z, Yan L, Liu Y, Yang H, Li H: Global, regional, and national cancer incidence and death for 29 cancer groups in 2019 and trends analysis of the global cancer burden, 1990–2019. J Hematol Oncol 2021, 14(1): 197.
- Ashrafi A, Akter Z, Modareszadeh P, Modareszadeh P, Berisha E, Alemi PS, Chacon Castro MdC, Deese AR, Zhang L: Current landscape of therapeutic resistance in lung cancer and promising strategies to overcome resistance. Cancers (Basel) 2022, 14(19): 4562.
- Gedye C, Navani V: Find the path of least resistance: Adaptive therapy to delay treatment failure and improve outcomes. Biochim Biophys Acta Rev Cancer 2022, 1877(2): 188681.
- Fhu CW, Ali A: Dysregulation of the ubiquitin proteasome system in human malignancies: a window for therapeutic intervention. Cancers (Basel) 2021, 13(7): 1513.
- Komander D, Clague MJ, Urbé S: Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009, 10(8): 550-563.
- Clague MJ, Urbé S, Komander D: Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol 2019, 20(6): 338-352.
- Zhang N, Zhang H, Yang X, Xue Q, Wang Q, Chang R, Zhu L, Chen Z, Liu X: USP14 exhibits high expression levels in hepatocellular carcinoma and plays a crucial role in promoting the growth of liver cancer cells through the HK2/AKT/P62 axis. BMC cancer 2024, 24(1): 237.
- Huang G, Li L, Zhou W: USP14 activation promotes tumor progression in hepatocellular carcinoma. Oncol Rep 2015, 34(6): 2917-2924.
- Chen M-y, Li Z-p, Sun Z-n, Ma M: USP9X promotes the progression of hepatocellular carcinoma by regulating beta-catenin. Ir J Med Sci (1971-) 2020, 189(3): 865-871.
- Yi T: Role of ubiquitin-specific proteases in hepatocellular carcinoma. Curr Top Med Chem 2025, 41(5): 968-974.
- Zhang X, Jin J, Cong J, Chen S, Wang T, Gao B, Huang G, Huang Z, Zhang J, Wang Z, et al: Role of ubiquitin-specific proteases in hepatocellular carcinoma pathogenesis. Curr Top Med Chem 2024, 24(3): 179-191.
- Xu X, Chen Y, Shao S, Wang J, Shan J, Wang Y, Wang Y, Chang J, Zhou T, Chen R, et al: USP21 deubiquitinates and stabilizes HSP90 and ENO1 to promote aerobic glycolysis and proliferation in cholangiocarcinoma. Int J Biol Sci 2024, 20(4): 1492.
- Zhang DY, Zhu Y, Wu Q, Ma S, Ma Y, Shen ZC, Wang Z, Sun W, Zhou YC, Wang D, et al: USP1 promotes cholangiocarcinoma progression by deubiquitinating PARP1 to prevent its proteasomal degradation. Cell Death Dis 2023, 14(10): 669.
- Chen Y, Xu X, Wang Y, Zhang Y, Zhou T, Jiang W, Wang Z, Chang J, Liu S, Chen R, et al: Hypoxia-induced SKA3 promoted cholangiocarcinoma progression and chemoresistance by enhancing fatty acid synthesis via the regulation of PAR-dependent HIF-1a deubiquitylation. J Exp Clin Cancer Res 2023, 42(1): 265.
- Li S, Song Y, Wang K, Liu G, Dong X, Yang F, Chen G, Cao C, Zhang H, Wang M: USP32 deubiquitinase: cellular functions, regulatory mechanisms, and potential as a cancer therapy target. Cell Death Discov 2023, 9(1): 338.
- Zhu S, Zhang X, Liu W, Zhou Z, Xiong S, Li J, Chen X, Peng C: Ubiquitination in cancer: mechanisms and therapeutic opportunities. Cancer Commun (Lond) 2025, Epub ahead of print.
- Goyal K, Afzal M, Bishoyi AK, Roopashree R, Saini S, Sharma R, Pathak PK, Chauhan AS, Aravindhan S, Imran M: Ubiquitin-specific proteases in hepatitis: bridging molecular mechanisms and therapeutic potential. Egypt Liver J 2025, 15(1): 1-18.
- Li Z, Duan D, Li L, Peng D, Ming Y, Ni R, Liu Y: Tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for hepatocellular carcinoma: recent research progress. Front Pharmacol 2024, 15: 1382256.
- Crew MA, Brennan TJ: Business models: Some implications for USPS. In: Postal and delivery innovation in the digital economy. Epub ahead of print, edn.: Springer; 2014, 1-15.
- Pohl C, Dikic I: Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366(6467): 818-822.
- Komander D, Rape M: The ubiquitin code. Annu Rev Biochem 2012, 81(1): 203-229.
- Swatek KN, Komander D: Ubiquitin modifications. Cell Res.2016, 26(4): 399-422.
- Harrigan JA, Jacq X, Martin NM, Jackson SP: Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov 2018, 17(1): 57-78.
- Antao AM, Tyagi A, Kim K-S, Ramakrishna S: Advances in deubiquitinating enzyme inhibition and applications in cancer therapeutics. Cancers 2020, 12(6): 1579.
- Mevissen TE, Komander D: Mechanisms of deubiquitinase specificity and regulation. Annu Rev Biochem 2017, 86(1): 159-192.
- Luo L, Sun W, Zhu W, Li S, Zhang W, Xu X, Fang D, Grahn THM, Jiang L, Zheng Y: BCAT1 decreases the sensitivity of cancer cells to cisplatin by regulating mTOR-mediated autophagy via branched-chain amino acid metabolism. Cell Death Dis 2021, 12(2): 169.
- Bakkar M, Khalil S, Bhayekar K, Kushwaha ND, Samarbakhsh A, Dorandish S, Edwards H, Dou QP, Ge Y, Gavande NS: Ubiquitin-Specific Protease Inhibitors for Cancer Therapy: Recent Advances and Future Prospects. Biomolecules 2025, 15(2): 240.
- Yang Y-C, Zhao C-J, Jin Z-F, Zheng J, Ma L-T: Targeted therapy based on ubiquitin-specific proteases, signalling pathways and E3 ligases in non-small-cell lung cancer. Front Oncol 2023, 13: 1120828.
- Pang L, Huang Y, Huang-Gao J, Chen P: Protease regulation of tumor-immune cell symbiosis. Trends Cancer 2025, 11(6): 560-574.
- Sun J, Zhu Z, Li W, Shen M, Cao C, Sun Q, Guo Z, Liu L, Wu D: UBE2T-regulated H2AX monoubiquitination induces hepatocellular carcinoma radioresistance by facilitating CHK1 activation. J Exp Clin Cancer Res 2020, 39(1): 222.
- Mi X, Li Q, Long G, Pan Y, Xie Y, Lu S, Xiao L, Tang J, Zhou L: Deubiquitylating Enzymes in Hepatocellular Carcinoma. Int J Biol Sci 2025, 21(9): 4270.
- Spataro V, Buetti-Dinh A: POH1/Rpn11/PSMD14: a journey from basic research in fission yeast to a prognostic marker and a druggable target in cancer cells. Br J Cancer 2022, 127(5): 788-799.
- Jee S-C, Cheong H: Autophagy/mitophagy regulated by ubiquitination: a promising pathway in cancer therapeutics. Cancers 2023, 15(4): 1112.
- Yuan J, Luo K, Zhang L, Cheville JC, Lou Z: USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140(3): 384-396.
- Czech-Sioli M, Siebels S, Radau S, Zahedi RP, Schmidt C, Dobner T, Grundhoff A, Fischer N: The ubiquitin-specific protease Usp7, a novel Merkel cell polyomavirus large T-antigen interaction partner, modulates viral DNA replication. J Virol 2020, 94(5): e01638-19.
- Zhang Q, Jia Q, Gao W, Zhang W: The role of deubiquitinases in virus replication and host innate immune response. Front Microbiol 2022, 13: 839624.
- Floreani A, Gabbia D, De Martin S: Current perspectives on the molecular and clinical relationships between primary biliary cholangitis and hepatocellular carcinoma. Int J Mol Sci 2024, 25(4): 2194.
- Gupta A, Satish A, Singh K, Saxena P: Gallbladder Carcinoma: A Comprehensive Review and Recent Updates. 2025, Epub ahead of print.
- Ye Z, Xie B, Tao Y, Xiao D: Mechanism of Ferroptosis and Its Role in Disease Development. Int J Biol Sci 2025, 21(12): 5328.
- Tu R, Ma J, Zhang P, Kang Y, Xiong X, Zhu J, Li M, Zhang C: The emerging role of deubiquitylating enzymes as therapeutic targets in cancer metabolism. Cancer Cell Int 2022, 22(1): 130.
- Zeng K, Xie W, Wang C, Wang S, Liu W, Su Y, Lin L, Zou R, Sun G, Zhou B: USP22 upregulates ZEB1-mediated VEGFA transcription in hepatocellular carcinoma. Cell Death Dis 2023, 14(3): 194.
- Xia D, Zhu X, Wang Y, Gong P, Su H-S, Xu X: Implications of ubiquitination and the maintenance of replication fork stability in cancer therapy. Biosci Rep 2023, 43(10): BSR20222591.
- Shen G, Lin Y, Yang X, Zhang J, Xu Z, Jia H: MicroRNA-26b inhibits epithelial-mesenchymal transition in hepatocellular carcinoma by targeting USP9X. BMC cancer 2014, 14(1): 393.
- Long G, Wu Z, Wang D, Mi X, Hu K, Zhou L, Tang J: UCHL3 inhibits ferroptosis by stabilizing β-catenin and maintains stem-like properties of hepatocellular carcinoma cells. Free Radic Biol Med 2024, 212: 162-173.
- Pal S, Sharma A, Mathew SP, Jaganathan BG: Targeting cancer-specific metabolic pathways for developing novel cancer therapeutics. Front Immunol 2022, 13: 955476.
- Ling S, Shan Q, Zhan Q, Ye Q, Liu P, Xu S, He X, Ma J, Xiang J, Jiang G, et al: USP22 promotes hypoxia-induced hepatocellular carcinoma stemness by a HIF1α/USP22 positive feedback loop upon TP53 inactivation. Gut 2020, 69(7): 1322-1334.
- Guo J, Zhao J: USP22-JMJD8 axis promotes Lenvatinib resistance in hepatocellular carcinoma. Biochim Biophys Acta Mol Cell Res 2024, 1871(1): 119617.
- Zhu H, Yan F, Yuan T, Qian M, Zhou T, Dai X, Cao J, Ying M, Dong X, He Q, et al: USP10 promotes proliferation of hepatocellular carcinoma by deubiquitinating and stabilizing YAP/TAZ. Cancer Res 2020, 80(11): 2204-2216.
- Ma Y-S, Chu K-J, Liu J-B, Cao P-S, Gu L-P, Fu D, Zhang X-W: Long Noncoding RNA OIP5-AS1 Promotes the Progression of Liver Hepatocellular Carcinoma via Regulating miR-26a-3p/EPHA2 Axis. Mol Ther Nucleic Acids 2020, 21: 229-241.
- Ma W, Zhang J, Chen W, Liu N, Wu T: The histone lysine acetyltransferase KAT2B inhibits cholangiocarcinoma growth: evidence for interaction with SP1 to regulate NF2-YAP signaling. J Exp Clin Cancer Res 2024, 43(1): 117.
- Cai B, Zhao J, Zhang Y, Liu Y, Ma C, Yi F, Zheng Y, Zhang L, Chen T, Liu H, et al: USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy 2022, 18(5): 990-1004.
- Aryapour E, Kietzmann T: Mitochondria, mitophagy, and the role of deubiquitinases as novel therapeutic targets in liver pathology. J Cell Biochem 2022, 123(10): 1634-1646.
- Liu C, Yuan Y, Zhan Y, Zou M, Wu L, Zhang C, Chen B, Zeng H, Yang R, Hu T, et al: Role of the USP family in autophagy regulation and cancer progression. Apoptosis 2025, 30(5-6): 1133-1151.
- Wu L, Fu Z, Zhou S, Gong J, Liu CA, Qiao Z, Li S: HIF-1α and HIF-2α: Siblings in promoting angiogenesis of residual hepatocellular carcinoma after high-intensity focused ultrasound ablation. PLoS One 2014, 9(2): e88913.
- Li X, Zhang Q, Wang Z, Zhuang Q, Zhao M: Immune and metabolic alterations in liver fibrosis: a disruption of oxygen homeostasis? Front Mol Biosci 2022, 8: 802251.
- Xiao S, Jiang S, Wen C, Wang H, Nie W, Zhao J, Zhang B: EMC2 promotes breast cancer progression and enhances sensitivity to PDK1/AKT inhibition by deubiquitinating ENO1. Int J Biol Sci 2025, 21(6): 2629.
- Xiao S, Tian L, Gan X, Xu X, Liao M, Song D, Yu Y, Qin W, Zhang R, Lyu H, et al: Role of Ubiquitin-regulated EMT in Cancer Metastasis and Chemoresistance. Int J Biol Sci 2025, 21(14): 6081-6112.
- Lv X-Y, Duan T, Li J: The multiple roles of deubiquitinases in liver cancer. Am J Cancer Res 2020, 10(6): 1647.
- Jia M, Guo Y, Lu X: USP33 is a biomarker of disease recurrence in papillary thyroid carcinoma. Cell Physiol Biochem 2018, 45(5): 2044-2053.
- Tao L, Liu X, Jiang X, Zhang K, Wang Y, Li X, Jiang S, Han T: USP10 as a potential therapeutic target in human cancers. Genes 2022, 13(5): 831.
- Zhang M, Wei T, Guo D: The role of abnormal ubiquitination in hepatocellular carcinoma pathology. Cell Signal 2024, 114: 110994.
- Yu J, Ling S, Hong J, Zhang L, Zhou W, Yin L, Xu S, Que Q, Wu Y, Zhan Q, et al: TP53/mTORC1-mediated bidirectional regulation of PD-L1 modulates immune evasion in hepatocellular carcinoma. J Immunother Cancer 2023, 11(11): e007479.
- Bayat M, Nahid-Samiei R, Sadri Nahand J, Naghili B: Interferon and immunity: the role of microRNA in viral evasion strategies. Front Immunol 2025, 16: 1567459.
- Chang Y-S, Su C-W, Chen S-C, Chen Y-Y, Liang Y-J, Wu J-C: Upregulation of USP22 and ABCC1 during sorafenib treatment of hepatocellular carcinoma contribute to development of resistance. Cells 2022, 11(4): 634.
- Yang X, Zang H, Luo Y, Wu J, Fang Z, Zhu W, Li Y: High expression of USP22 predicts poor prognosis and advanced clinicopathological features in solid tumors: a meta-analysis. Onco Targets Ther 2018, 11: 3035-3046.
- Sonego M, Pellarin I, Costa A, Vinciguerra GLR, Coan M, Kraut A, D’andrea S, Dall’Acqua A, Castillo-Tong DC, Califano D, et al: USP1 links platinum resistance to cancer cell dissemination by regulating Snail stability. Sci Adv 2019, 5(5): eaav3235.
- Chen R, Zhang H, Li L, Li J, Xie J, Weng J, Tan H, Liu Y, Guo T, Wang M, et al: Roles of ubiquitin-specific proteases in inflammatory diseases. Front Immunol 2024, 15: 1258740.
- Kaushal K, Ramakrishna S: Deubiquitinating enzyme-mediated signaling networks in cancer stem cells. Cancers 2020, 12(11): 3253.
- Wang L, Wang J, Ma X, Ju G, Shi C, Wang W, Wu J: USP35 promotes HCC development by stabilizing ABHD17C and activating the PI3K/AKT signaling pathway. Cell Death Discov 2023, 9(1): 421.
- Shan Q, Yin L, Zhan Q, Yu J, Pan S, Zhuo J, Zhou W, Bao J, Zhang L, Hong J, et al: The p-MYH9/USP22/HIF-1α axis promotes lenvatinib resistance and cancer stemness in hepatocellular carcinoma. Signal Transduct Target Ther 2024, 9(1): 249.
- Ghosh S, Saha T: Central role of ubiquitination in genome maintenance: DNA replication and damage repair. ISRN Mol Biol 2012, 2012(1): 146748.
- Kwon S-K, Saindane M, Baek K-H: p53 stability is regulated by diverse deubiquitinating enzymes. Biochim Biophys Acta Rev Cancer 2017, 1868(2): 404-411.
- Wang Y, Li S, Wang W: The ubiquitin-proteasome system in the tumor immune microenvironment: a key force in combination therapy. Front Immunol 2024, 15: 1436174.
- Liu J, Leung CT, Liang L, Wang Y, Chen J, Lai KP, Tse WKF: Deubiquitinases in cancers: aspects of proliferation, metastasis, and apoptosis. Cancers 2022, 14(14): 3547.
- Ren J, Yu P, Liu S, Li R, Niu X, Chen Y, Zhang Z, Zhou F, Zhang L: Deubiquitylating enzymes in cancer and immunity. Adv Sci (Weinh) 2023, 10(36): 2303807.
- Zhang H, Liu W, Wu Y, Chen C: USP3: Key deubiquitylation enzyme in human diseases. Cancer Sci 2024, 115(7): 2094-2106.
- Tu R, Kang W, Yang M, Wang L, Bao Q, Chen Z, Dong Y, Wang J, Jiang J, Liu H, et al: USP29 coordinates MYC and HIF1α stabilization to promote tumor metabolism and progression. Oncogene 2021, 40(46): 6417-6429.
- Moya-Guzmán MJ, de Solminihac J, Padilla C, Rojas C, Pinto C, Himmel T, Pino-Lagos K: Extracellular vesicles from immune cells: A biomedical perspective. Int J Mol Sci 2023, 24(18): 13775.
- Hsu S-K, Kuo I-Y, Lin P-Y, Chou C-K, Ko C-C, Chang W-T, Chiu C-C: Deubiquitinating enzymes: Key regulators of ferroptosis and pyroptosis and novel targets for cancer intervention. Int J Mol Sci 2025, 21(9): 3993.
- Arimoto K-i, Miyauchi S, Troutman TD, Zhang Y, Liu M, Stoner SA, Davis AG, Fan J-B, Huang Y-J, Yan M, et al: Expansion of interferon inducible gene pool via USP18 inhibition promotes cancer cell pyroptosis. Nat Commun 2023, 14(1): 251.
- Zhang L, Wang Z, Liu K, Liu Y, Wang S, Jiang W, Lu F, Dang Y: Targets of tumor microenvironment for potential drug development. MedComm (2020) 2024, 3(1): e68.
- Jiang Y, Yu Z, Wang J, Wu Y, Li Z, Le T, Yang C, Wei Y, Zhang G, Ma H, et al: Targeting USP47 enhances immunotherapy in hepatocellular carcinoma by destabilizing PD-L1. IInt Immunopharmacol 2025, 161: 115024.
- Qian G, Zhu L, Li G, Liu Y, Zhang Z, Pan J, Lv H: An integrated view of deubiquitinating enzymes involved in type I interferon signaling, host defense and antiviral activities. Front Immunol 2021, 12: 742542.
- Ning Z, Wang A, Liang J, Xie Y, Liu J, Yan Q, Wang Z: USP22 promotes epithelial-mesenchymal transition via the FAK pathway in pancreatic cancer cells. Oncol Rep 2014, 32(4): 1451-1458.
- Xue Y, Xue C, Song W: Emerging roles of deubiquitinating enzymes in actin cytoskeleton and tumor metastasis. Cell Oncol 2024, 47(4): 1071-1089.
- Kalathil D, John S, Nair AS: FOXM1 and cancer: faulty cellular signaling derails homeostasis. Front Oncol 2021, 10: 626836.
- Marin JJ, Macias RI, Monte MJ, Romero MR, Asensio M, Sanchez-Martin A, Cives-Losada C, Temprano AG, Espinosa-Escudero R, Reviejo M, et al: Molecular bases of drug resistance in hepatocellular carcinoma. Cancers 2020, 12(6): 1663.
- Bansal N, Kumar A, Sharma P, Anikhandi S, Khare S, Arora A: Precision Medicine in Hepatobiliary Diseases: A Clinical Review. J Intern Med 2024, 5: 21-32.
- Pantsar T: Designing effective lead molecules against solid tumors with inadequate treatment options, the efficacy problem. 2018, Epub ahead of print.
- Qi S-M, Cheng G, Cheng X-D, Xu Z, Xu B, Zhang W-D, Qin J-J: Targeting USP7-mediated deubiquitination of MDM2/MDMX-p53 pathway for cancer therapy: are we there yet? Front Cell Dev Biol 2020, 8: 233.
- Ren X, Wang L, Liu L, Liu J: PTMs of PD-1/PD-L1 and PROTACs application for improving cancer immunotherapy. Front Immunol 2024, 15: 1392546.
- Zhao J, Guo J, Wang Y, Ma Q, Shi Y, Cheng F, Lu Q, Fu W, Ouyang G, Zhang J, et al: Research progress of DUB enzyme in hepatocellular carcinoma. Front Oncol 2022, 12: 920287.
- Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, Zhang Q, Chen J, Ott CA, Sun H, et al: A selective USP1–UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol 2014, 10(4): 298-304.
- Lanzafame M, Bianco G, Terracciano LM, Ng CK, Piscuoglio S: The role of long non-coding RNAs in hepatocarcinogenesis. Int J Mol Sci 2018, 19(3): 682.
- Wang X, Su Y, Lan B, Li X, Zhang B, Zhang L, Wang Y, Zhang C, Xuan C: USP22 promotes the proliferation and Sorafenib resistance of hepatocellular carcinoma cells via its deubiquitinase activity. Clin Transl Med 2025, 15(5): e70324.
- Murgai A, Sosič I, Gobec M, Lemnitzer P, Proj M, Wittenburg S, Voget R, Gütschow M, Krönke J, Steinebach C, et al: Targeting the deubiquitinase USP7 for degradation with PROTACs. Chem Commun (Camb) 2022, 58(63): 8858-8861.
- Antonenko S, Zavelevich M, Telegeev G: The role of USP1 deubiquitinase in the pathogenesis and therapy of cancer. Acta Biochim Pol 2023, 70(2): 219-231.
- Zhang X, Linder S, Bazzaro M: Drug development targeting the ubiquitin–proteasome system (UPS) for the treatment of human cancers. Cancers 2020, 12(4): 902.
- Gao H, Yin J, Ji C, Yu X, Xue J, Guan X, Zhang S, Liu X, Xing F: Targeting ubiquitin specific proteases (USPs) in cancer immunotherapy: from basic research to preclinical application. J Exp Clin Cancer Res 2023, 42(1): 225.
- Palmer DH, Johnson PJ: Evaluating the role of treatment-related toxicities in the challenges facing targeted therapies for advanced hepatocellular carcinoma. Cancer Metastasis Rev 2015, 34(3): 497-509.
- Hu X, Wu Y, Yao M, Chen Z, Li Q: The other side of the coin: protein deubiquitination by Ubiquitin-Specific Protease 1 in cancer progression and therapy. Future Med Chem 2025, 17(3): 329-345.
- Jena R, Samal HB, Sharma J, Suresh P, Mishra AP, Nigam M: Biotechnology in Drug Discovery and Development for Cancer. In: Biotechnology and Cancer Therapeutics. Epub ahead of print., edn.: Springer; 2025, 447-478.
- Kitamura H: Ubiquitin-specific proteases (USPs) and metabolic disorders. Int J Mol Sci 2023, 24(4): 3219.
- Lu J, Zhao H, Yu C, Kang Y, Yang X: Targeting ubiquitin-specific protease 7 (USP7) in cancer: a new insight to overcome drug resistance. Front Pharmacol 2021, 12: 648491.
- Gavali S, Liu J, Li X, Paolino M: Ubiquitination in T-cell activation and checkpoint inhibition: new avenues for targeted cancer immunotherapy. Int J Mol Sci 2021, 22(19): 10800.
- An T, Lu Y, Yan X, Hou J: Insights into the properties, biological functions, and regulation of USP21. Front Pharmacol 2022, 13: 944089.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.