
 Submit Manuscript
Submit Manuscript
Research Article | Open Access
Whole-exome sequencing reveals genetic underpinnings of tongue carcinoma in Chinese population
Shuhang Wang1*, Ning Jiang1*, Zicheng Yu2*, Yuan Fang1*, Shujun Xing1, Huiyao Huang1, Yue Yu1, Qi Fan1, Chao Zhang2, Zaixian Tai2, Jiucheng Liu2, Ruixian He3, Ning Li11Clinical Trial Center, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 100021, Beijing China.
2GenePlus-Shenzhen, 518000, Shenzhen China.
3Nursing Department, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 100021, Beijing China.
Correspondence: Ning Li (Clinical Trial Center, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 100021, Beijing China; E-mail: Lining@cicams.ac.cn). Ruixian He (Nursing Department, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 100021, Beijing China; E-mail: He87787591@126.com).
Asia-Pacific Journal of Oncology 2020, 1: 52-60. https://doi.org/10.32948/ajo.2020.10.15
Received: 17 Sep 2020 | Accepted: 10 Oct 2020 | Published online: 19 Oct 2020
Oral tongue squamous cell carcinoma (OTSCC) is a common malignancy, of which the incidence has increased in China in the last decade. Surprisingly, while multiple studies have revealed the mutational features of OTSCC in Western populations, limited data was shown in Asian patients. Herein, we utilized whole-exome sequencing to profile the genetic alterations in 13 Chinese OTSCC and compared them to those from 40 Western patients published in Cancer Discovery. In result, some key driver mutations were observed in both Chinese and Western cohorts, such as TP53 (Chinese 60.0% vs Western 60.0%), FAT1 (Chinese 7.7% vs Western 30.0%), CASP8 (Chinese 7.7% vs Western 10.0%) and NOTCH1 (Chinese 15.4% vs Western 10.0%), while mutations in CDKN2A (23.1%) and NTRK3 (23.1%) were only observed in Chinese patients, indicating these two novel mutations might play vital roles in OTSCC tumorigenesis specifically in Asian population. Mutational signatures depicted both common and distinct features across cohorts. In addition, significant copy number loss was found in 7q22.1, 9q13.1, and focal regions spanning CDKN2A and CDKN2B. FOXP1-TEX261 (2p13.3:3p13) fusion, reported in various cancer types, was firstly observed in OTSCC. Also, we identified numerous actionable mutations with FDA approved targeted. Taken together, our study revealed the mutational features of Chinese OTSCC patients, either similar or distinct to those of Caucasian patients. CDKN2A and NTRK3 were observed as two novel drivers that might play essential roles in tumorigenesis in Chinese patients, and were found as two potential therapeutic targets, rendering it promising to develop novel therapies.
Key words Actionable mutation, copy number alteration, oral tongue squamous cell carcinoma, whole-exome sequencing
13 patients diagnosed with OTSCC were enrolled from Cancer Hospital Chinese Academy of Medical Sciences from 2012 to 2019. Detailed clinicopathological characteristics of all patients were shown in Table S1. All patients underwent resection. Primary tumor tissues together with matched normal specimens were collected surgically and snap frozen in liquid nitrogen and stored at -80. DNA extraction was performed with TIANamp Genomic DNA Kits (Tiangen Biotech, Beijing, China) according to manufacturer’s instructions. Written informed consent was collected in sample collection, gene sequencing and data publication. This study was approved by the Institutional Review Board (IRB) of Cancer Hospital Chinese Academy of Medical Sciences and conducted in accordance with the Declaration of Helsinki.
Library Construction and Whole-exome Sequencing
Library construction was performed using a custom 53M length capturing probe, made by Integrated DNA Technologies (IDT, IA, USA). Captured libraries were then pair-end sequenced in 100-bp lengths with Geneplus-2000 sequencing platform (Geneplus, Beijing, China) following the manufacturer’s guidance. Reads were further mapped to the reference human genome (hg19) utilizing BWA aligner (version 0.7.10) for mutation calling after filtration.
Mutation Calling
Somatic single nucleotide variants (SNVs), small insertions and deletions (Indels) were detected using MuTect (version 1.1.4) packed in GATK (version 4.0). Variants with high frequencies among unrelated individuals across the world or specifically in east Asia were firstly filtered out as previously reported [6] and were listed in Table S2, then further filtered with the method demonstrated in a study [7] in order to screen out the key cancer drivers or cancer associated mutations. In brief, candidate alterations had to be mutations in known cancer associated genes collected in either Catalogue of Somatic Mutations in Cancer (COMSIC), OncoKB, CIvic (Clinical Interpretation of Variants in Cancer), CGI (Cancer Genome Interpreter) or IntOGEN (Integrative Onco-Genomics) database (all accessed in July, 2020). Mutations including nonsense, in-frame/frame-shift small insertions and deletions, and mutations in canonical splice-sites were kept. In regard of missense mutations, only those reported in COSMIC with a FATHMM-MKL (Functional Analysis through Hidden Markov Models) score of >0.5 (accessed in August 2020), or those identified in two or more of the following in silico functional analysis algorithms: predication score 0-0.05 in SIFT (Sorting Intolerant from Tolerant), “possibly damaging” or “probably damaging” in Polymorphism Phenotyping-2 (Polyphen2) or predication >0.5 in FATHMM-MKL were kept.
To study the significantly altered copy number of chromosomal segments, somatic copy number alterations (SCNA) was identified employing GATK (version 4.0) and GISTIC (version 2.0). An in-house algorithm was employed in detecting split reads and discordant read-pairs to identify chromosomal structural variation [6, 8]. Eventually, all candidate somatic variations were manually verified on the integrative genomics reviewer (IGV) browser to filter out the false positive.
To compare the mutational features between Chinese and Caucasian OTSCC populations, mutation data from WES of 40 Caucasian OTSCC patients, published in 2013 on Cancer Discovery [5] were downloaded from cBioPortal for Cancer Genomics database, with detailed clinicopathological information exhibited in Table S1. Mutations in genes collected in either COSMIC, OncoKB, CIvic, CGI or IntOGEN database were kept and subjected to following analyses. To find potential therapeutic targets, we mapped the unfiltered raw SNVs and Indels to OncoKB actionable mutation catalogue (accessed in July 2020), so as to avoid missing any potentially valuable candidates.
Analysis of Mutational Signature and Clonal Architecture
To find the possible mechanisms underlying OTSCC mutagenesis and reason the potential carcinogen-exposure or DNA damage repair deficiency of patients, the trinucleotide altering patterns of unfiltered somatic SNVs and fragment changing patterns of unfiltered Indels were matched to the known signatures described in COSMIC database using YAPSA (version 3.11). Contribution of each signature in samples was quantified and only signatures with contributions of more than 1% of all alterations were illustrated and considered in the subsequent analyses. Clonality of mutational events were inferred by looking at the variant allele frequency (VAF) ratio of mutations, calculated in the way as previously reported [9]. Specifically, VAF ratio was calculated by dividing the VAF of each mutation by the maximum VAF observed in the same sample. A higher VAF ratio suggested the respective event occurred at an earlier stage during tumor progression. Mutations with VAF ratios > 0.75 were determined as clonal mutations while the rest were considered as subclonal mutations.
Statistics
Two-sided Mann-Whitney and Fisher’s exact tests were performed on Graphpad Prism (version 7.01) or R (version 3.6.1) to generate the P value. All data processing, analyses and plotting were carried out utilizing R software (version 3.6.1) and/or GraphPad Prism software (version 8.0.2). Statistical significance was defined as P<0.05 for two-tailed student t-tests.
Whole-exome sequencing identified a total of 1,456 somatic SNVs and Indels in 13 Chinese OTSCC patients (CHN cohort) before filtration, while 3, 471 mutations in 40 Caucasian patients published in Cancer Discovery (CD cohort) were reported [5]. After filtration, 235 and 536 mutations in known cancer-associated genes were identified in CHN and CD cohorts respectively. In CHN cohort, genes with the highest mutation frequencies were TP53 (11/13, 84.6%), CDKN2A (3/13, 23.1%) and NTRK3 (3/13, 23.1%) (Figure 1A). Other recurrently mutated genes included ARHGAP5, CHD6, CNTNAP2, CSMD3, FAM47C, FAT4, GRIN2A, HCFC1, HERC2, KDM3B, KIAA1549, MUC4, NOTCH1, NOTCH2, ROBO4, SETBP1 and SETD1B, all mutated in 2/13 patients (15.4%) (Figure 1A). Mapping mutations into Reactome (accessed in September 2020) database suggested that mutations were enriched in NOTCH signaling pathway, RUNX3 signaling pathway, VENTX pathway and pathways involved in cellular senescence, which are closely related to cell cycle, apoptosis and stem cell phenotypes, and were shown to be associated with tumorigenesis in various cancer types [10-13]. In regard of CD cohort, recurrently mutated genes included TP53 (24/40, 60%), FAT1 (12/40, 30%), MUC4 (10/40, 25%), NEFH (7/40, 17.5%), PCLO (6/40, 15%), PIK3CA (6/40, 15%), HRAS (5/40, 12.5%), HLA-B (5/40, 12.5%), MLL2 (5/40, 12.5%), CASP8 (4/40, 10%), CHEK2 (4/40, 10%), HLA-A (4/40, 10%), MUC16 (4/40, 10%), NOTCH1 (4/40, 10%), SMAD4 (4/40, 10%) and ZNF521 (4/40, 10%) (Figure 1A).
We then compared the frequencies of mutations in known cancer-associated genes between CHN and CD cohorts (Figure 1B). In result, we observed similar mutational frequencies in some canonical cancer driver genes between two cohorts, such as TP53 (CD 60% vs. CHN 60%), FAT1 (CD 30% vs. CHN 7.7%), CASP8 (CD 10% vs. CHN 7.7%) and NOTCH1 (CD 10% vs. CHN 15.4%), indicating that they might play pivotal roles in OTSCC tumorigenesis. Notably, mutation frequencies of CDKN2A and NTRK3 in our cohort (3/13, 23.0% for both) were observed significantly higher than in CD cohort (0 for both; P = 0.012 for both; Figure 1B), suggesting CDKN2A and NTRK3 might be two novel drivers in OTSCC cancer progression among Chinese population.
To further justify the association of CDKN2A and NTRK3 with OTSCC tumorigenesis, we looked at the locations of their altered nucleotides on the protein-annotated chromosomes accordingly. As shown in (Figure 1C & Figure 1D), 2/3 missense mutations in NTRK3 located in the functional domains, namely immunoglobulin I-set domain and protein tyrosine kinase domain, whereas another mutation located in a non-functional region. As for CDKN2A mutations, 2/3 truncating nonsense mutations located in ankyrin repeats domain while another mutation located at a canonical splice site.
Mutational Signatures of Somatic SNVs and Indels in OTSCC
To gain an insight into OTSCC cancer etiology, we then matched the altering patterns of raw somatic SNVs and Indels to COSMIC single base signatures (SBS) and insertion and deletion signatures (ID) accordingly and quantified the contribution of each signature in both CHN cohort and CD cohort. In result, somatic SNVs were mainly attributed to three SBSs in COSMIC database, SBS39, SBS7b, SBS1 specifically, while Indels were attributed to ID1-14, ID16 and ID17 (Figure 2A).
SBS39, featured with predominant C > G alterations, was observed with drastically high distribution in head and squamous cell carcinoma and breast cancer and without known proposed etiology [14]. SBS7b, characterized by C > T mutations, was frequently found in skin cancer and was reported to be due to exposure to ultraviolet light [14]. SBS1, a clock-like signature with predominant C > T changes, is an endogenous mutational process initiated by spontaneous or enzymatic deamination of 5-methylcytosine to thymine which generates G: T mismatches in double-strand DNA, and was consistent with previous reports that SBS1 was one of the main signatures of OTSCC [15]. In our cohort, ID5 was observed particularly predominant, characterized by predominant single T base deletion, and was reported to be a clock-like signature associated with patients’ ages [14].
We further quantified and compared the contribution of identified SBSs and IDs in CHN and CD cohorts to look at the difference in possible mutagenic exposure and DNA damage repair deficiency between Chinese and Caucasian populations. Interestingly, the exposing contribution of SBS1 was found significantly lower in our cohort compared to CD cohort (P < 0.001; Figure 2B), whereas SBS7b and ID5 were both observed with substantially increased contribution in our cohort (P < 0.001 for both) (Figure 2C & Figure 2D).
Clonal Architecture of Somatic Mutations in OTSCC
To look at the intra-tumor heterogeneity and infer the temporal order of the occurrence of somatic SNVs and Indels in Chinese OTSCC, we gauged the variant allele frequency (VAF) ratio of all alterations in our cohort with the method mentioned in previous literature [9]. Somatic mutations with higher VAF ratios were suggested to occur at earlier stages during tumor progression [9]. Mutations with VAF ratios greater than 0.5 were exhibited in (Figure 3), where black dots represent median VAF ratios when multiple mutations in the same genes were identified. Being consistent with the findings stated above, mutations in canonical cancer driver genes such as TP53 (0.78), CDKN2A (0.82) and CSMD3 (0.81) were determined as clonal mutations with VAF ratios greater than 0.75, indicating they might occur at an earlier stage compared to other subclonal mutations, and might play pivotal roles in OTSCC tumorigenesis. By contrast, some other canonical cancer drivers like KRAS, FAT1, NOTCH1 and APC were observed with relatively lower VAF ratios, suggesting they might occur at a later stage in tumor progression, exerting functions in tumor development and subclonal evolution. Subclonal mutations with various VAF ratios largely contributed to OTSCC intra-tumor heterogeneity and might be able to prime tumors towards drug resistant, disease relapse and metastasis.
Somatic Copy Number Alteration in OTSCC
To identify the recurrent focal somatic copy number alterations (SCNA), the GISTIC algorithm and the 2Mb windows setting were applied to our cohort. Among population, two significant regions of deletion, including 7q22.1 and 9q13.1, were detected (Figure 4A), while no significant amplified large segment was identified (Figure 4B).
We also manually checked the smaller focal regions of amplification and deletion of individual patients in order to characterize the driver genes significantly implicated in copy number changes. As a result, canonical oncogenes such as MYC, CCND1, FGFR1, ERBB2, PDGFRA, and KIT were gained or amplified whereas tumor suppressor genes such as CDKN2A, CDKN2B, and BRCA1 were lost or deleted (Table S3).
Chromosomal Variation in OTSCC
A total of 28 structural changes were identified in our cohort (10/13, 76.9%). Of particular interest, among 10 OTSCC patients harboring chromosomal changes, one case was paid special attention to and discussed in detail due to the detection of cancer associated FOXP1-TEX261 (2p13.3:3p13) fusion (Figure 5), which might exert potential functions in enticing tumorigenesis and were previously reported in other cancer types encompassing aldosterone-producing adenoma [16], prostate cancer acute lymphoplastic leukemia and etc [17-19].
Actionable Mutations of OTSCC in Chinese Population
In order to develop novel therapeutic interventions for OTSCC, we looked at the actionable mutations in our cohort by mapping unfiltered raw mutations into OncoKB actionable mutation catalogue (Figure 6), so that all potential candidate targets could be kept. Multiple actionable genes were observed with recurrent mutations, including CDKN2A, NTRK3, FGFR1, FGFR2, CCND1, BRCA1 and etc, functioning as potential candidates for developing novel targeted therapies. Of particular interest, CDKN2A and NTRK3 were observed mutated with the highest prevalence in Chinese cohort whereas no mutation in these genes was found in Caucasian cohort, suggesting they might play essential roles in OTSCC tumorigenesis and could be considered as two promising therapeutic targets, specifically in Asian population.
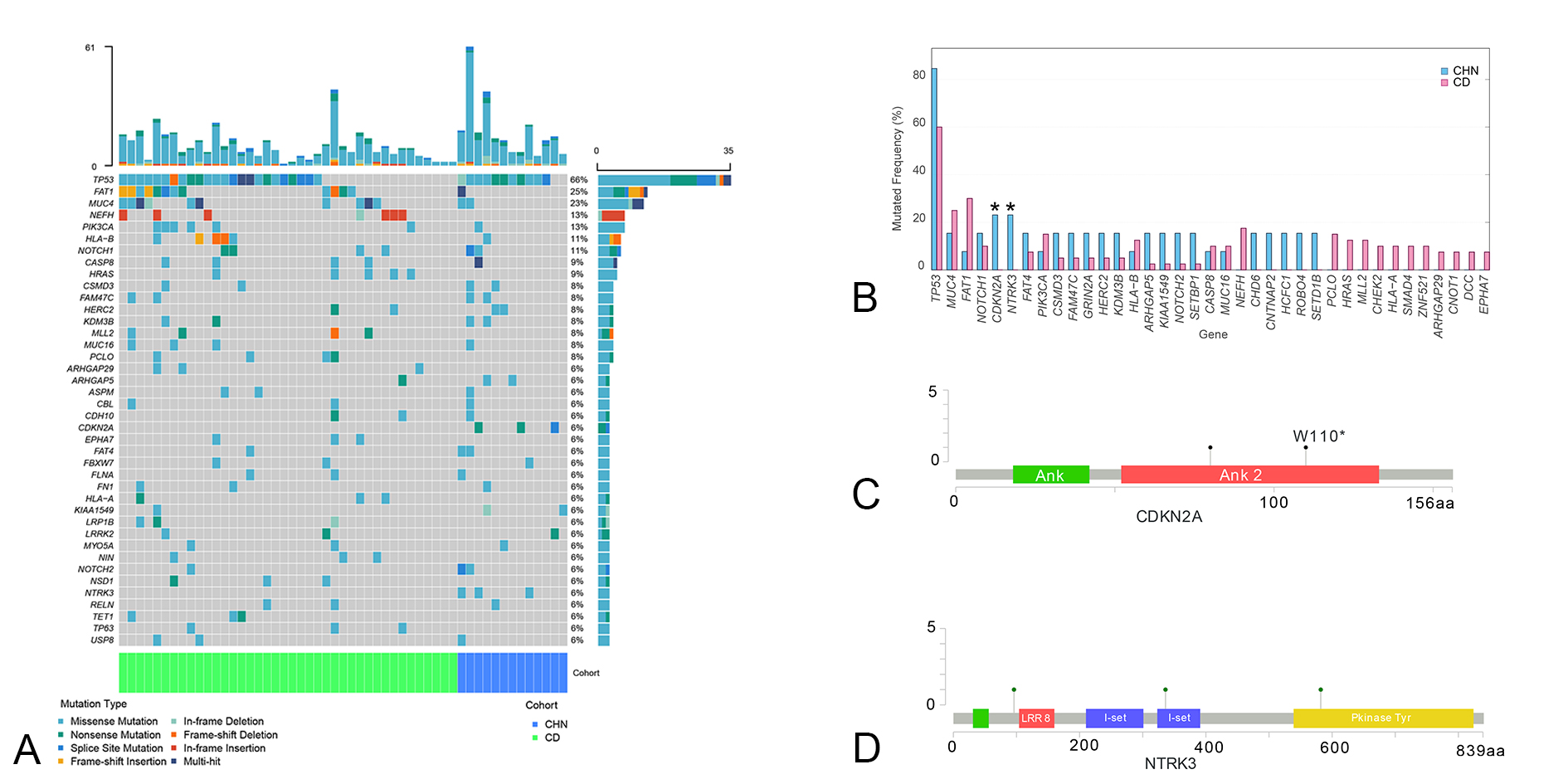 Figure 1. Somatic Single Nucleotide Variations and Small Insertions and Deletions Detected; A. Landscape of all mutations identified, panel at the top, mutation counts in each patient, right panel, number of patients harboring mutations in specific genes, panel at the bottom, source of patients, CHN- Chinese patients in our cohort (N = 13); CD-Patients published in Cancer Discovery (N = 40); B. Prevalence of Mutations in each cohort, * represents a P value < 0.05; C. Locations of sites of CDKN2A somatic SNVs/Indels in our cohort, Ank - ankyrin repeats domain; D. Locations of sites of NTRK3 somatic SNVs/Indels in our cohort, LRR8 - Leucine-rich repeat domain; I-set - Immunoglobulin I-set domain; Pkinase Tyr - Tyrosine protein kinases specific active-site signature domain.
Figure 1. Somatic Single Nucleotide Variations and Small Insertions and Deletions Detected; A. Landscape of all mutations identified, panel at the top, mutation counts in each patient, right panel, number of patients harboring mutations in specific genes, panel at the bottom, source of patients, CHN- Chinese patients in our cohort (N = 13); CD-Patients published in Cancer Discovery (N = 40); B. Prevalence of Mutations in each cohort, * represents a P value < 0.05; C. Locations of sites of CDKN2A somatic SNVs/Indels in our cohort, Ank - ankyrin repeats domain; D. Locations of sites of NTRK3 somatic SNVs/Indels in our cohort, LRR8 - Leucine-rich repeat domain; I-set - Immunoglobulin I-set domain; Pkinase Tyr - Tyrosine protein kinases specific active-site signature domain.
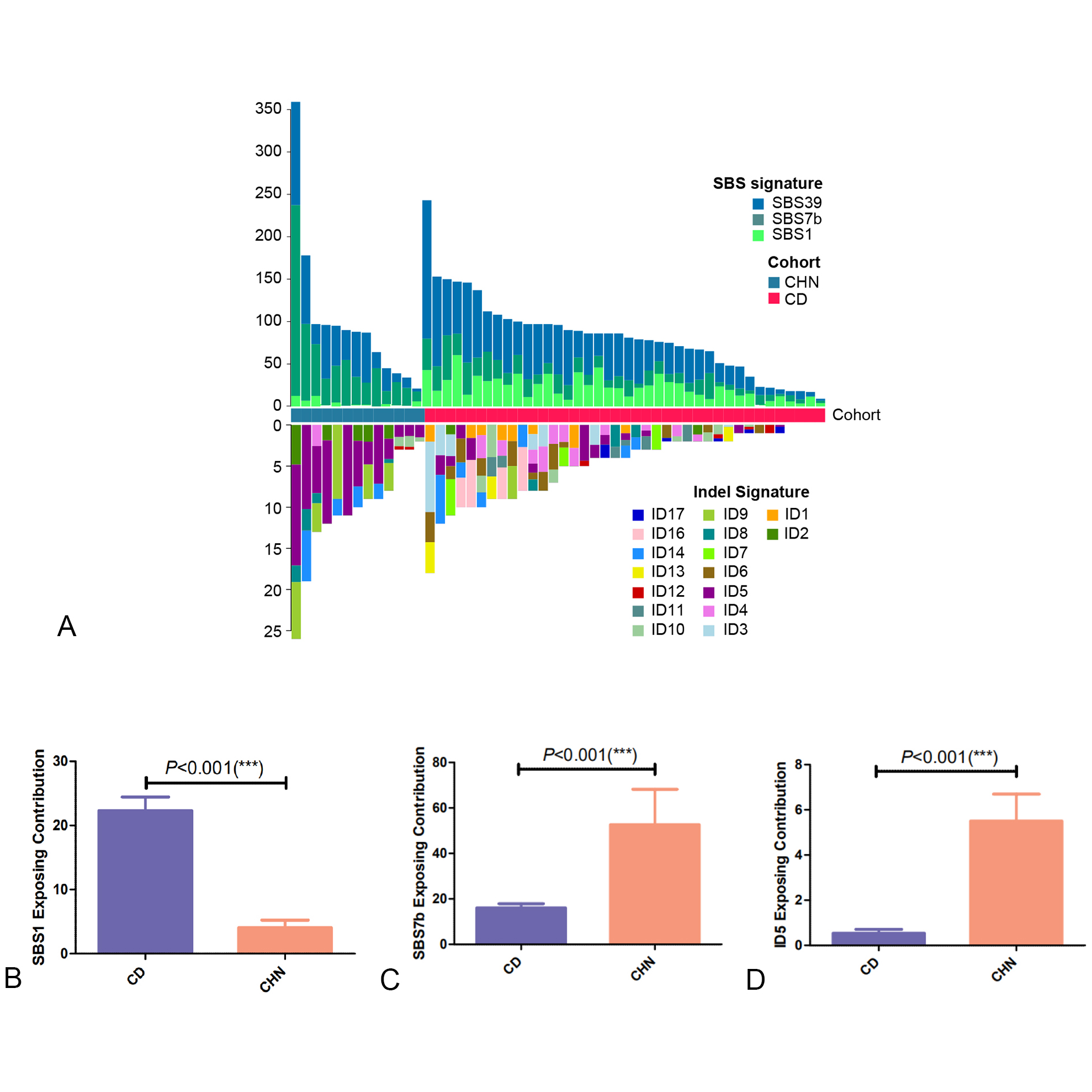 Figure 2. Exposing Contribution of COSMIC signatures in 13 Chinese OTSCC patients; A. panel at the top, exposing contribution of SBS signatures; panel in the middle, source of patients, CHN- Chinese patients in our cohort (N = 13), CD-Patients published in Cancer Discovery (N = 40); panel at the bottom, exposing contribution of Indel signatures; B, C, D. exposing contribution of SBS1, SBS7b, ID5 in CHN vs. CD cohorts accordingly.
Figure 2. Exposing Contribution of COSMIC signatures in 13 Chinese OTSCC patients; A. panel at the top, exposing contribution of SBS signatures; panel in the middle, source of patients, CHN- Chinese patients in our cohort (N = 13), CD-Patients published in Cancer Discovery (N = 40); panel at the bottom, exposing contribution of Indel signatures; B, C, D. exposing contribution of SBS1, SBS7b, ID5 in CHN vs. CD cohorts accordingly.
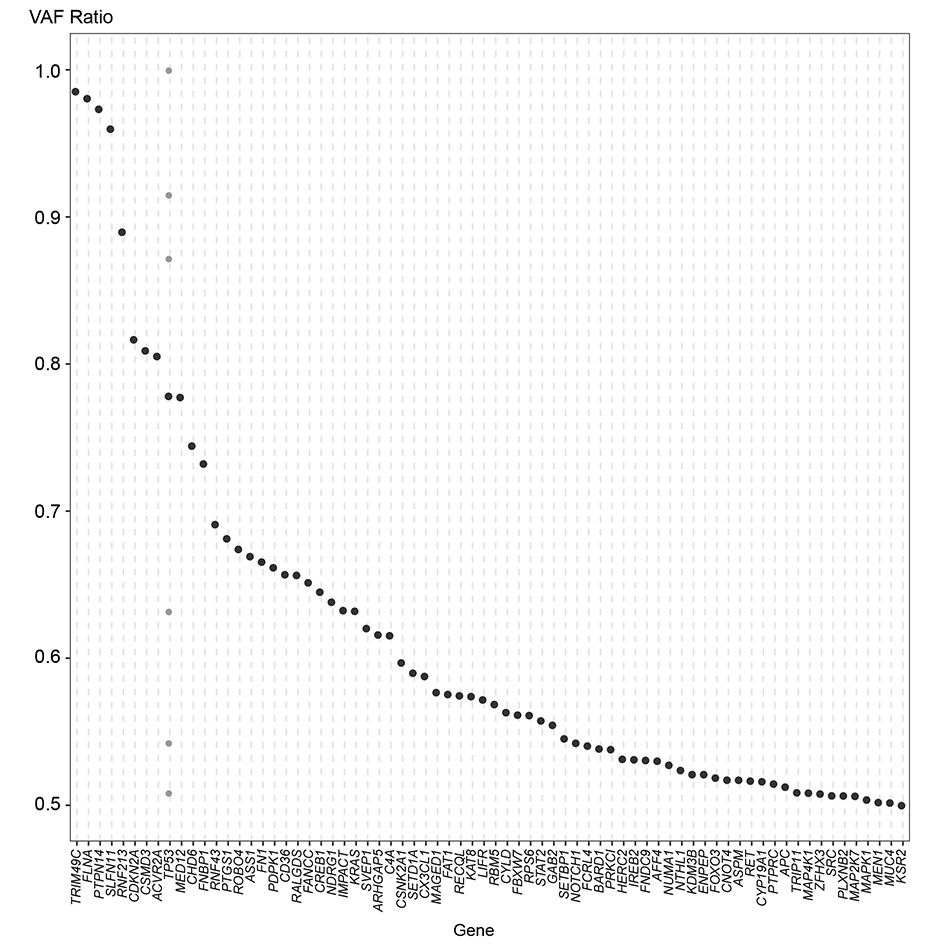 Figure 3. Mutations in genes with VAF ratios equals to or larger than 0.5 in 13 OTSCC patients.
Figure 3. Mutations in genes with VAF ratios equals to or larger than 0.5 in 13 OTSCC patients.
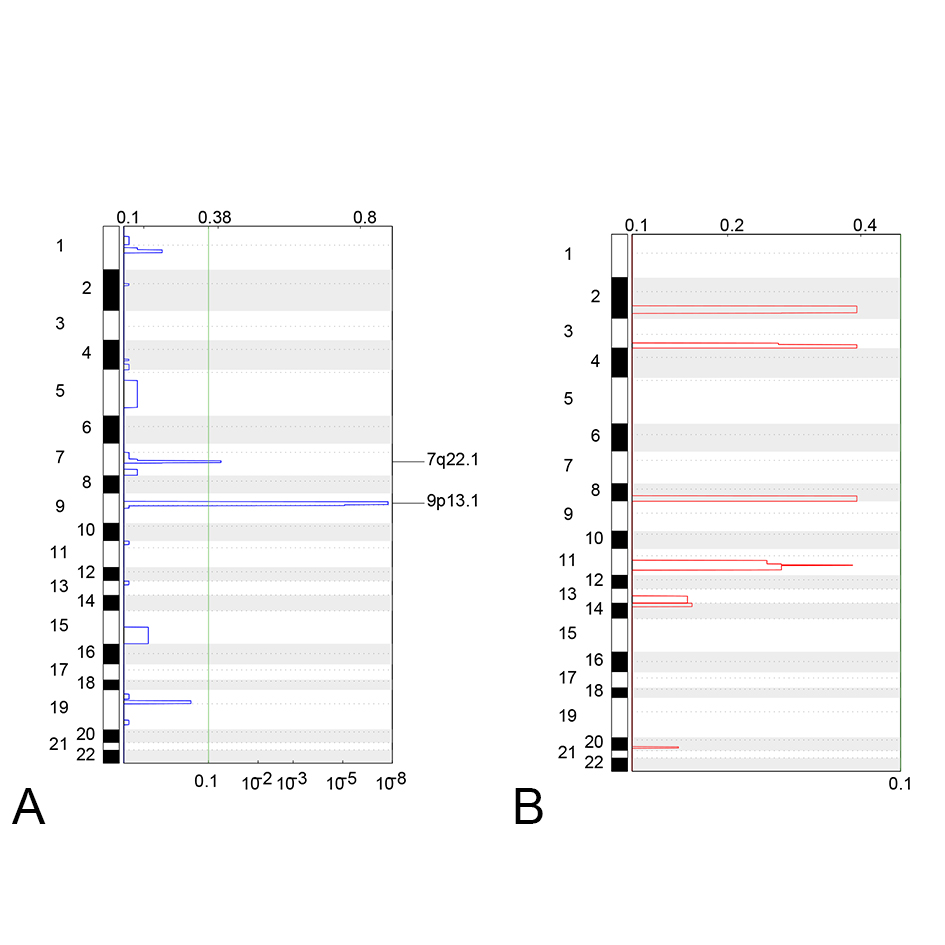 Figure 4. Significant loss (A) or Gain (B) of copy number segments in 13 OTSCC patients. On each figure, the panel in the left represents the chromosome of each CNV segment located on; the numbers on the top represents the G-scores assigned by GISTIC for every cytoband plotted; the numbers on the bottom represents the -log10 transformed q values of each CNV.
Figure 4. Significant loss (A) or Gain (B) of copy number segments in 13 OTSCC patients. On each figure, the panel in the left represents the chromosome of each CNV segment located on; the numbers on the top represents the G-scores assigned by GISTIC for every cytoband plotted; the numbers on the bottom represents the -log10 transformed q values of each CNV.
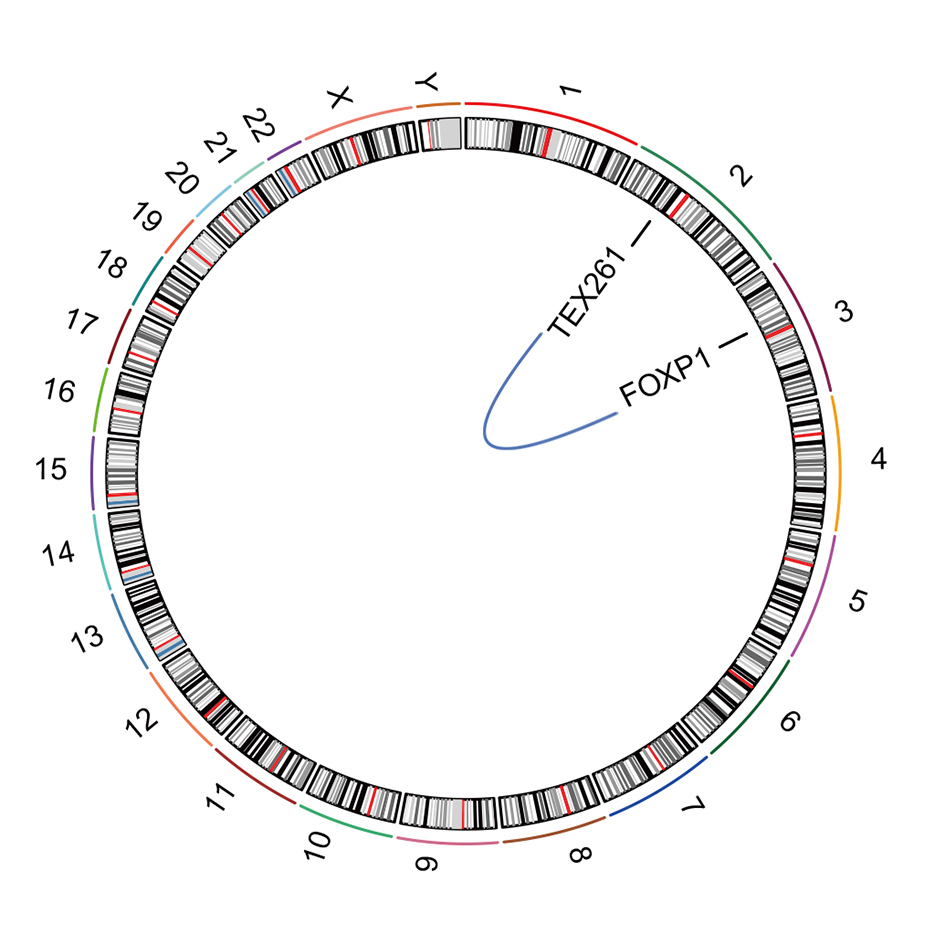 Figure 5. TEX261-FOXP1 fusion identified in 13 OTSCC patients.
Figure 5. TEX261-FOXP1 fusion identified in 13 OTSCC patients.
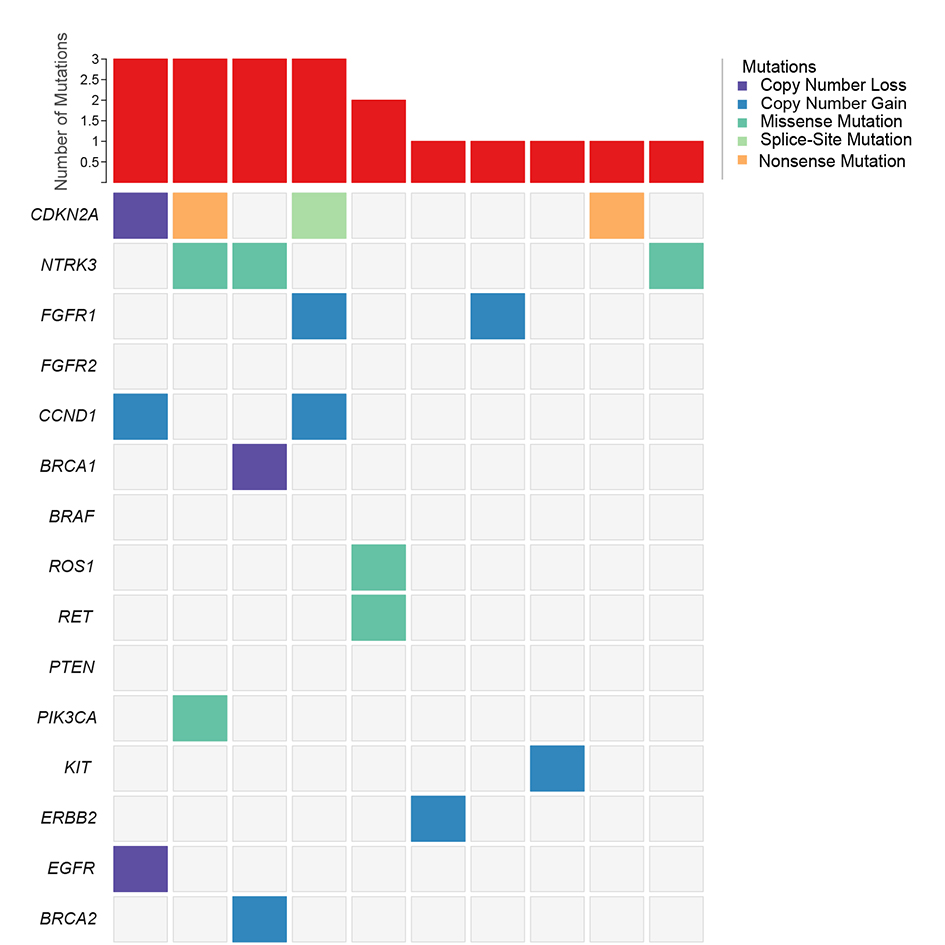 Figure 6. Mutations in Actionable Mutations by Searching OncoKB actionable genes in raw mutations of 13 Chinese OTSCC patients.
Figure 6. Mutations in Actionable Mutations by Searching OncoKB actionable genes in raw mutations of 13 Chinese OTSCC patients.
Overall, compared to Western patients, the mutation frequencies of some known cancer associated genes in Chinese patients were quite close [5]. To this end, some key mutations in oncogenes and tumor suppressor genes such as TP53, PIK3CA, CASP8, NOTCH1, MUC4, FAT1 and etc were identified in both our cohort and CD cohort, proving their roles in OTSCC tumorigenesis as previously reported [5]. Also, our results suggested that the pathways these genes enriched in, NOTCH signaling pathway and CASP8 cellular necrosis pathway for instance, might be engaged in OTSCC cancer progression.
Notably, mutations in CDKN2A and NTRK3, both occurred in 23.1% patients in our cohort, were instead undetected in CD cohort, and both were categorized in OncoKB database as actionable genes, potentially offering novel targets for therapeutic development. Previously, CDKN2A and NTRK3 were reported to play key roles in various cancer types in the Chinese population including non-small cell lung cancer [21]. CDKN2A is a canonical tumor suppressor gene encoding p16INK4a protein, and was demonstrated to be a key driver triggering tumorigenesis in various cancer types [8, 22, 23]. The encoded p16INK4a protein exerts functions to orchestrate multiple signaling pathways in modulating cell cycle and senescence, through regulating the cyclin-dependent kinase (CDK) 4/6 and cyclin D complexes [8, 22, 23]. Moreover, genetic and epigenetic alterations in CDKN2A were exhibited to be indicative of disease relapse and dismal prognosis in cancer patients [8, 22, 23]. In our cohort, 2 truncating nonsense mutations and 1 mutation in canonical splice site were identified in 13 patients, and both 2 nonsense mutations located in the functional ankyrin repeats domain, which was shown to be involved in protein-protein interaction of p16INK4a to constrain cells from passing through cell cycle, and was suggested to play key roles in p16INK4a function [24, 25]. Our result indicated that mutations in CDKN2A might lead to the loss of p16INK4a regulatory capacity in modulating cell cycle arrest, and further result in OTSCC tumorigenesis. In addition, clonal CDKN2A mutation with high VAF ratio detected in our cohort suggested that CDKN2A might be altered at an early stage compared to other genes with subclonal mutations. Significant copy number loss of CDKN2A together with CDKN2B were also found recurrent in our cohort, which potentially resulted in a decreased expression level of p16 proteins, reducing its ability to maintain normal cell cycle arrest. Similarly, a recent study showed that CDKN2A copy number loss was observed highly prevalent (146/401, 36.4%) in HPV-negative head and neck squamous cell carcinoma, stringently associated with patients’ dismal prognosis [26]. A germline polymorphism CDKN2B-AS1 (rs2151280 T>C) was also shown by another study to be associated with a higher risk of lung cancer [27]. Besides, differential expression in tumor tissues was found in various cancer types including hepatoblastoma, colorectal cancer and pituitary adenomas etc [28-31]. The down-regulation of CDKN2A and CDKN2B were found to associate with the recurrence of disease in oral cancer patients [32]. In 2003, Yakushiji et al showed that by comparing 25 pairs of primary oral squamous cell carcinoma (OSCC) and matched normal oral mucosa tissues, hypermethylation and down-regulation in both mRNA and protein expression levels were found in 48% samples, demonstrating a mechanism of CDKN2A undergoing hypermethylation, down-regulation to eventually trigger OSCC tumorigenesis [33]. In-vitro knock-in and knock-down assays of CDKN2A exhibited its basic functions in triggering G1 phase arrest during cell cycle in cancer cell-lines, where CDKN2A down-regulation was shown to be associated with enhanced cell proliferation [34, 35]. Besides, a CRISPR-Cas9 engineered CDKN2A knock-out model demonstrated depletion of CDKN2A facilitated melanocyte in-vitro motility and in-vivo invasiveness, indicating the function of CDKN2A to inhibit E2F1-mediated transcriptional activation of BRN2, which in turn to regulate down-stream signaling cascades to trigger melanoma metastasis [36]. Taken together, these results further solidified the conjecture that CDKN2A might be a novel cancer driver gene in OTSCC. The discrepancy of CDKN2A mutation in our cohort compared to CD cohort might be due to modest sample sizes of these two studies, while another possible explanation is somatic CDKN2A mutations might be specific to Asian OTSCC population whereas they occur less frequently in Caucasian population, awaiting more substantiation.
NTRK3 is another cancer suppressor gene regulating cell survival and apoptosis, encoding neurotrophin-3 receptor, which functions as a tyrosine kinase receptor to orchestrate multiple signal transduction cascades involved in cellular proliferation and was demonstrated to play important roles in various cancer types [37, 38]. Similarly, NTRK3 was found differentially expressed in various cancer types. Methylation and down-regulation were observed in tumors where reconstitution induced cell apoptosis, and a decreased expression of NTRK3 was reported to be associated with neoplastic transformation, cell migration and invasion in-vitro and in-vivo [39-42]. Aberrantly methylated NTRK3 was shown to be a biomarker of colorectal cancer [43]. Down-regulation of NTRK3 was suggested to increase cancer cell invasion, migration and proliferation [42, 44]. Moreover, ETV6-NTRK3 fusion was reported to be driver alteration triggering tumorigenesis of multiple cancer types, including secretory breast cancer, colorectal cancer and Spitz tumor [37, 45-48]. Recently, NTRK somatic mutations and ETV6-NTRK3 fusion were also reported in head and neck squamous cell cancers with a prevalence of 7.9% and 2.4% in Chinese patients (3/127; 10/127) respectively, and one of the ETV6-NTRK3 positive patient benefited from crizotinib treatment, offering an alternative treatment strategy [49]. Similar to CDKN2A, NTRK3 was observed recurrently mutated in our cohort, while no mutation was detected in CD cohort. 2 missense mutations of NTRK3 were found located in the immunoglobulin I-set domain and protein tyrosine kinase domain, leading to NTRK3 loss of function. Therefore, NTRK3 might also be another novel driver facilitating tumorigenesis and tumor development in OTSCC, and possibly specific to Asian population. However, ETV6-NTRK3 fusion was not detected in our cohort, possibly due to a limited sample size and/or a different sequencing strategy utilized in our study.
From our result, 76.9% OTSCC harbored mutations sensitive to off-the-shelf targeted therapeutic drugs, rendering targeted therapy a highly promising means of intervention to this malignancy. We manually checked the FDA approved medications in OncoKB database (Memorial Sloan Kettering Cancer Center) for these candidate genes. Among which, gefitinib, afatinib, osimertinib and dacomitinib were approved for EGFR+ non-small cell lung cancer, rucaparib was approved for BRCA1+ ovarian cancer, sunitinib, imatinib and regorafenib were approved for KIT+ gastrointestinal stromal tumor, and fulvestrant with alpelisib was approved for PIK3CA+ breast cancer etc. It’s worth noting that even though these drugs were approved in numerous cancer types, limited numbers of drugs were developed or approved to be applied to OTSCC to date, therefore developing new therapeutic strategies for this cancer type might be of great significance to improve patients’ treatment opportunities.
The mutational signatures of oral cavity cancer appear to vary depending on the exposure of environmental carcinogens [50], as habitual consumption of tobacco, alcohol, and betel quid (BQ) were recognized as the major etiologic factors for this type of malignancies. Previous literature has depicted the distinctive mutational features in tumors associated with cigarette smoking [51], alcohol drinking [52], and BQ chewing [53, 54]. In present study, we conducted a quantitative measurement of mutational patterns for our cohort and CD cohort. In result, though consistent signatures were identified between our cohort with previous report, significant difference was observed in contribution of mutational signatures between two cohorts. The discrepancy in signature distribution is considered reasonable given that the living environment and lifestyle largely vary across the populations.
Copy number alteration analysis revealed the deleterious somatic SCNA in hallmark genes of tongue cancer, such as amplification of MYC, CCND1 and FGFR1, and were also identified in previous reports [16, 55, 56]. In addition, high expression of ERBB2 was reported to exhibit important roles in the development of tongue cancer in patients with smoking history and/or alcohol consumption [57], and PDGFRA expression was significantly higher in oral cancer cohort with or without the establishment of tobacco risk factor [58]. Also as stated above, the down-regulation of CDKN2A and CDKN2B were found to associate with the recurrence of disease in oral cancer patients [32]. In conclusion, our SCNA findings were consistent with previous literature and have vital implications in clinically relevant applications.
Despite no overall structural variation pattern scrutinized, multiple chromosomal changes occurred in individuals might potentially contribute to cancer progression. In one patient, a FOXP1-TEX261 (2p13.3:3p13) fusion was observed (Figure 6). FOXP1 (Forkhead Box P1) encodes transcription factor Foxp1 expressed across the whole system, and was previously reported to exert functions in regulating immune response, cancer progression, organ development, and showed oncogene properties in acute lymphoblastic leukemia [19, 59-62]. As chromosomal changes involving FOXP1 was frequently found in various malignancies, FOXP1 served as a highly potential gene target for developing therapeutic interventions [62].
The study was supported by the funder Chinese Academy of Medical Sciences(2019XK320068), and Chinese Academy of Medical Sciences(2020-I2M-2-007).
Author contributions
NL designed and supervised the study, RXH collect and preserve the patient samples. SHW, JN, ZY and YF analyzed the data and composed the article. SX collected clinical data and samples. YY, QF, CZ, ZXT, JL verified the data and methods. All the authors have read the article and were involved in the review and revision.
Competing interests
The authors declare that they have no conflicts of interest in this study.
Ethical statement
The study was approved by the Ethic Committee of Cancer Hospital Chinese Academy of Medical Sciences (ID: NCC-002760).
Additional files
Additional file 1: Table S1. Clinicopathological Characteristics of Patients Enrolled and in External Datasets.
Additional file 2: Table S2. Raw Mutations Identified from 13 Chinese OTSCC patients.
Additional file 3: Table S3. Significantly Implicated Driver Genes in Focal CNV Segments.
- Prince S, Bailey BM: Squamous carcinoma of the tongue: review. Br J Oral Maxillofac Surg 1999, 37(3): 164-174.
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018, 68(6): 394-424.
- Vettore AL, Ramnarayanan K, Poore G, Lim K, Ong CK, Huang KK, Leong HS, Chong FT, Lim TK, Lim WK, et al: Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med 2015, 7(1): 98.
- Su SC, Chang LC, Lin CW, Chen MK, Yu CP, Chung WH, Yang SF: Mutational signatures and mutagenic impacts associated with betel quid chewing in oral squamous cell carcinoma. Hum Genet 2019, 138(11-12): 1379-1389.
- Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, et al: Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov 2013, 3(7): 770-781.
- Li X, Huang H, Guan Y, Gong Y, He CY, Yi X, Qi M, Chen ZY: Whole-exome sequencing predicted cancer epitope trees of 23 early cervical cancers in Chinese women. Cancer Med 2017, 6(1): 207-219.
- Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, Nik-Zainal S, Steenbruggen TG, Tjan-Heijnen VCG, Labots M, et al: The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 2019, 51(10): 1450-1458.
- Bartsch DK, Sina-Frey M, Lang S, Wild A, Gerdes B, Barth P, Kress R, Grützmann R, Colombo-Benkmann M, Ziegler A, et al: CDKN2A germline mutations in familial pancreatic cancer. Ann Surg 2002, 236(6): 730-737.
- Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, et al: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012, 366(10): 883-892.
- Allenspach EJ, Maillard I, Aster JC, Pear WS: Notch signaling in cancer. Cancer Biol Ther 2002, 1(5): 466-476.
- Ito K, Liu Q, Salto-Tellez M, Yano T, Tada K, Ida H, Huang C, Shah N, Inoue M, Rajnakova A, et al: RUNX3, a novel tumor suppressor, is frequently inactivated in gastric cancer by protein mislocalization. Cancer Res 2005, 65(17): 7743-7750.
- Gao H, Le Y, Wu X, Silberstein LE, Giese RW, Zhu Z: VentX, a novel lymphoid-enhancing factor/T-cell factor-associated transcription repressor, is a putative tumor suppressor. Cancer Res 2010, 70(1): 202-211.
- Collado M, Blasco MA, Serrano M: Cellular senescence in cancer and aging. Cell 2007, 130(2): 223-233.
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al: The repertoire of mutational signatures in human cancer. Nature 2020, 578(7793): 94-101.
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN: The repertoire of mutational signatures in human cancer. BioRxiv 2018 Epub ahead of print.: 322859.
- Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun 2013, 4: 2873.
- Takayama K, Suzuki T, Tsutsumi S, Fujimura T, Takahashi S, Homma Y, Urano T, Aburatani H, Inoue S: Integrative analysis of FOXP1 function reveals a tumor-suppressive effect in prostate cancer. Mol Endocrinol 2014, 28(12): 2012-2024.
- Ernst T, Score J, Deininger M, Hidalgo-Curtis C, Lackie P, Ershler WB, Goldman JM, Cross NC, Grand F: Identification of FOXP1 and SNX2 as novel ABL1 fusion partners in acute lymphoblastic leukaemia. Br J Haematol 2011, 153(1): 43-46.
- Sugimoto Y, Sada A, Shimokariya Y, Monma F, Ohishi K, Masuya M, Nobori T, Matsui T, Katayama N: A novel FOXP1-PDGFRA fusion gene in myeloproliferative neoplasm with eosinophilia. Cancer Genet 2015, 208(10): 508-512.
- Chaturvedi AK, Engels EA, Anderson WF, Gillison ML: Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol 2008, 26(4): 612-619.
- Wen S, Dai L, Wang L, Wang W, Wu D, Wang K, He Z, Wang A, Chen H, Zhang P, et al: Genomic Signature of Driver Genes Identified by Target Next-Generation Sequencing in Chinese Non-Small Cell Lung Cancer. Oncologist 2019, 24(11): e1070-e1081.
- Foulkes WD, Flanders TY, Pollock PM, Hayward NK: The CDKN2A (p16) gene and human cancer. Mol Med 1997, 3(1): 5-20.
- Zhao R, Choi BY, Lee MH, Bode AM, Dong Z: Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016, 8: 30-39.
- Borg A, Sandberg T, Nilsson K, Johannsson O, Klinker M, Måsbäck A, Westerdahl J, Olsson H, Ingvar C: High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst 2000, 92(15): 1260-1266.
- Monzon J, Liu L, Brill H, Goldstein AM, Tucker MA, From L, McLaughlin J, Hogg D, Lassam NJ: CDKN2A mutations in multiple primary melanomas. N Engl J Med 1998, 338(13): 879-887.
- Chen WS, Bindra RS, Mo A, Hayman T, Husain Z, Contessa JN, Gaffney SG, Townsend JP, Yu JB: CDKN2A Copy Number Loss Is an Independent Prognostic Factor in HPV-Negative Head and Neck Squamous Cell Carcinoma. Front Oncol 2018, 8: 95.
- Lv X, Cui Z, Li H, Li J, Yang Z, Bi Y, Gao M, Zhang Z, Wang S, Zhou B, et al: Association between polymorphism in CDKN2B-AS1 gene and its interaction with smoking on the risk of lung cancer in a Chinese population. Hum Genomics 2019, 13(1): 58.
- Al-Grawi EDC, Al-Awsi GRL: Expression of CDKN2A (P16/Ink4a) among colorectal cancer patients: a cohort study. J Pharm Sci Res 2018, 10(5): 1145-1147.
- Guled M, Lahti L, Lindholm PM, Salmenkivi K, Bagwan I, Nicholson AG, Knuutila S: CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma -A miRNA microarray analysis. Genes Chromosomes Cancer 2009, 48(7): 615-623.
- Iolascon A, Giordani L, Moretti A, Basso G, Borriello A, Della Ragione F: Analysis of CDKN2A, CDKN2B, CDKN2C, and cyclin Ds gene status in hepatoblastoma. Hepatology 1998, 27(4): 989-995.
- Simpson DJ, Bicknell JE, McNicol AM, Clayton RN, Farrell WE: Hypermethylation of the p16/CDKN2A/MTSI gene and loss of protein expression is associated with nonfunctional pituitary adenomas but not somatotrophinomas. Genes Chromosomes Cancer 1999, 24(4): 328-336.
- Deepak Roshan VG, Sinto MS, Vargees BT, Kannan S: Loss of CDKN2A and CDKN2B expression is associated with disease recurrence in oral cancer. J Oral Maxillofac Pathol 2019, 23(1): 82-89.
- Yakushiji T, Uzawa K, Shibahara T, Noma H, Tanzawa H: Over-expression of DNA methyltransferases and CDKN2A gene methylation status in squamous cell carcinoma of the oral cavity. Int J Oncol 2003, 22(6): 1201-1207.
- Agarwal P, Sandey M, DeInnocentes P, Bird RC: Tumor suppressor gene p16/INK4A/CDKN2A-dependent regulation into and out of the cell cycle in a spontaneous canine model of breast cancer. J Cell Biochem 2013, 114(6): 1355-1363.
- Frost SJ, Simpson DJ, Clayton RN, Farrell WE: Transfection of an inducible p16/CDKN2A construct mediates reversible growth inhibition and G1 arrest in the AtT20 pituitary tumor cell line. Mol Endocrinol 1999, 13(11): 1801-1810.
- Zeng H, Jorapur A, Shain AH, Lang UE, Torres R, Zhang Y, McNeal AS, Botton T, Lin J, Donne M, et al: Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation. Cancer Cell 2018, 34(1): 56-68.e9.
- Wood LD, Calhoun ES, Silliman N, Ptak J, Szabo S, Powell SM, Riggins GJ, Wang TL, Yan H, Gazdar A, et al: Somatic mutations of GUCY2F, EPHA3, and NTRK3 in human cancers. Hum Mutat 2006, 27(10): 1060-1061.
- Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH: A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998, 18(2): 184-187.
- Bu JY, Lv WZ, Liao YF, Xiao XY, Lv BJ: RETRACTED: Long non-coding RNA LINC00978 promotes cell proliferation and tumorigenesis via regulating microRNA-497/NTRK3 axis in gastric cancer. Int J Biol Macromol 2019, 123: 1106-1114.
- Ge L, Li N, Liu M, Xu NZ, Wang MR, Wu LY: Copy number variations of neurotrophic tyrosine receptor kinase 3 (NTRK3) may predict prognosis of ovarian cancer. Medicine (Baltimore) 2017, 96(30): e7621.
- Luo Y, Kaz AM, Kanngurn S, Welsch P, Morris SM, Wang J, Lutterbaugh JD, Markowitz SD, Grady WM: NTRK3 is a potential tumor suppressor gene commonly inactivated by epigenetic mechanisms in colorectal cancer. PLoS Genet 2013, 9(7): e1003552.
- Xiong D, Sheng Y, Ding S, Chen J, Tan X, Zeng T, Qin D, Zhu L, Huang A, Tang H: LINC00052 regulates the expression of NTRK3 by miR-128 and miR-485-3p to strengthen HCC cells invasion and migration. Oncotarget 2016, 7(30): 47593-47608.
- Luo Y, Kaz AM, Kanngurn S, Welsch P, Morris SM, Wang J, Lutterbaugh JD, Markowitz SD, Grady WM: NTRK3 is a potential tumor suppressor gene commonly inactivated by epigenetic mechanisms in colorectal cancer. PLoS Genet 2013, 9(7): e1003552.
- Tu J, Zhao Z, Xu M, Chen M, Weng Q, Ji J: LINC00460 promotes hepatocellular carcinoma development through sponging miR-485-5p to up-regulate PAK1. Biomed Pharmacother 2019, 118: 109213.
- Li Z, Tognon CE, Godinho FJ, Yasaitis L, Hock H, Herschkowitz JI, Lannon CL, Cho E, Kim SJ, Bronson RT, et al: ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer Cell 2007, 12(6): 542-558.
- Euhus DM, Timmons CF, Tomlinson GE: ETV6-NTRK3--Trk-ing the primary event in human secretory breast cancer. Cancer Cell 2002, 2(5): 347-348.
- Yeh I, Tee MK, Botton T, Shain AH, Sparatta AJ, Gagnon A, Vemula SS, Garrido MC, Nakamaru K, Isoyama T, et al: NTRK3 kinase fusions in Spitz tumours. J Pathol 2016, 240(3): 282-290.
- Lambros MB, Tan DS, Jones RL, Vatcheva R, Savage K, Tamber N, Fenwick K, Mackay A, Ashworth A, Reis-Filho JS: Genomic profile of a secretory breast cancer with an ETV6-NTRK3 duplication. J Clin Pathol 2009, 62(7): 604-612.
- Xu J, Wang R, Wang T, Wang T, Shu Y, Gu D, He Y, Chen R, Liu L: The Genomic Profiling in Chinese Head and Neck Cancer and Incidence of NTRK Fusion. J Clin Pathol 2020 Epub ahead of print.
- Beck TN, Golemis EA: Genomic insights into head and neck cancer. Cancers Head Neck 2016, 1: 1.
- Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al: Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354(6312): 618-622.
- Chang J, Tan W, Ling Z, Xi R, Shao M, Chen M, Luo Y, Zhao Y, Liu Y, Huang X, et al: Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat Commun 2017, 8: 15290.
- Zhang W, Wang M, Wu Q, Zhu Q, Jiao Y, Zhu Y, Yang B, Ni S, Yu J, Sun H, et al: Mutational signatures and the genomic landscape of betel quid chewing-associated tongue carcinoma. Cancer Med 2019, 8(2): 701-711.
- Su SC, Chang LC, Lin CW, Chen MK, Yu CP, Chung WH, Yang SF: Mutational signatures and mutagenic impacts associated with betel quid chewing in oral squamous cell carcinoma. Hum Genet 2019, 138(11-12): 1379-1389.
- Upadhyay P, Gardi N, Desai S, Chandrani P, Joshi A, Dharavath B, Arora P, Bal M, Nair S, Dutt A: Genomic characterization of tobacco/nut chewing HPV-negative early stage tongue tumors identify MMP10 asa candidate to predict metastases. Oral Oncol 2017, 73: 56-64.
- Hammerman PS, Hayes DN, Grandis JR: Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov 2015, 5(3): 239-244.
- Zhao Y, Fu D, Xu C, Yang J, Wang Z: Identification of genes associated with tongue cancer in patients with a history of tobacco and/or alcohol use. Oncol Lett 2017, 13(2): 629-638.
- Ong HS, Gokavarapu S, Tian Z, Li J, Xu Q, Cao W, Zhang CP: PDGFRA mRNA is overexpressed in oral cancer patients as compared to normal subjects with a significant trend of overexpression among tobacco users. J Oral Pathol Med 2017, 46(8): 591-597.
- Barrans SL, Fenton JA, Banham A, Owen RG, Jack AS: Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood 2004, 104(9): 2933-2935.
- Streubel B, Vinatzer U, Lamprecht A, Raderer M, Chott A: T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia 2005, 19(4): 652-658.
- Carlsson P, Mahlapuu M: Forkhead transcription factors: key players in development and metabolism. Dev Biol 2002, 250(1): 1-23.
- Koon HB, Ippolito GC, Banham AH, Tucker PW: FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets 2007, 11(7): 955-965.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.