
 Submit Manuscript
Submit Manuscript
Review | Open Access
Molecular mechanism of tumor microenvironment and recent approaches for the immunotherapy of pancreatic cancer
Ayesha Fazal Nawaz1, Muhammad Arif1
1Department of Life Sciences, University of Trieste, Via Licio Giorgieri, 5, 34127 Trieste, Italy.
Correspondence: Muhammad Arif (Department of Life Sciences, University of Trieste, Via Licio Giorgieri, 5, 34127 Trieste, Italy; Email: arifbiotech144@gmail.com).
Asia-Pacific Journal of Oncology 2024, 5: 95-103. https://doi.org/10.32948/ajo.2024.12.10
Received: 15 Nov 2024 | Accepted: 10 Dec 2024 | Published online: 21 Dec 2024 
Key words pancreatic cancer, vaccine therapy, antibody therapy, cellular therapy, molecular mechanism
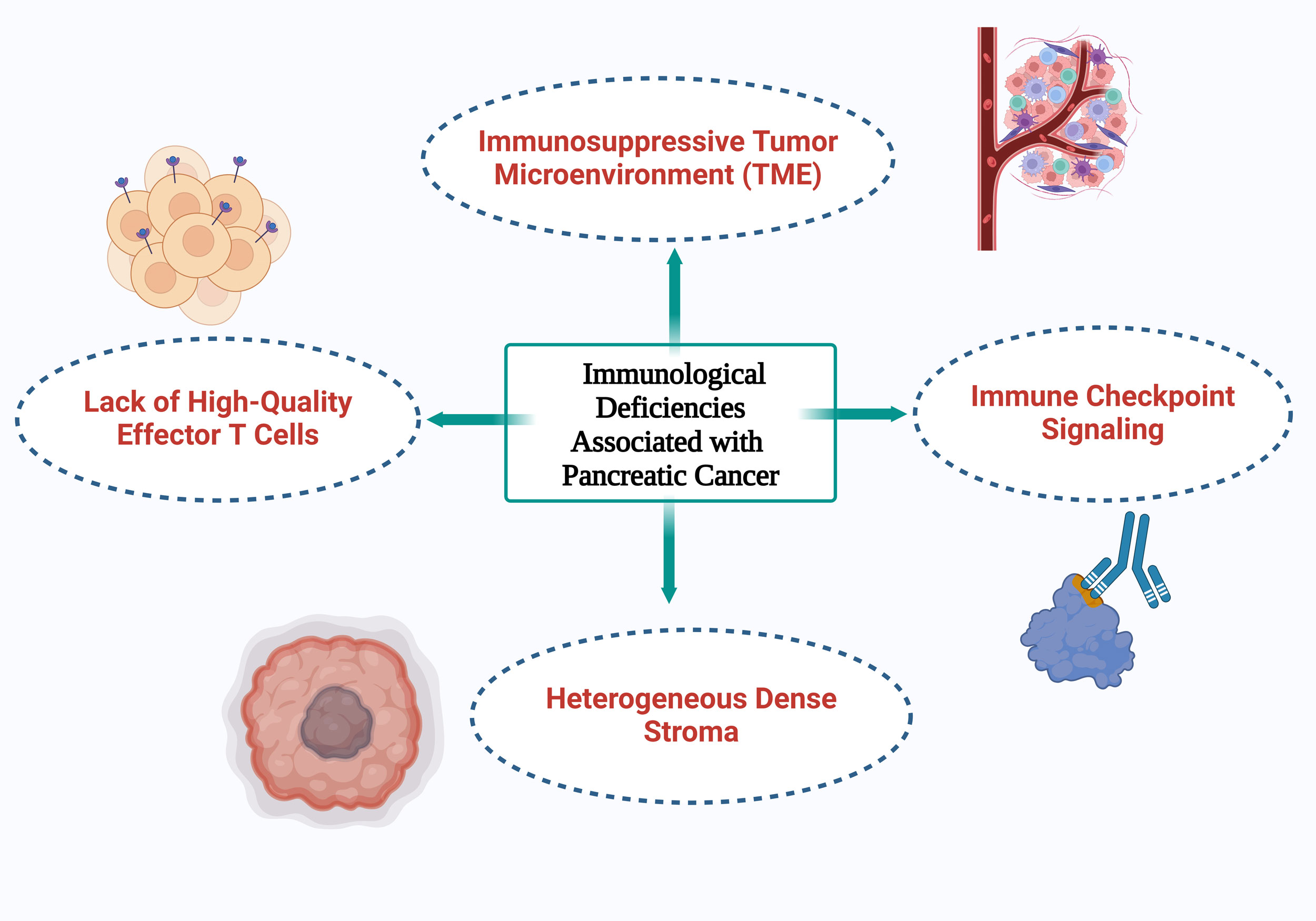 Figure 1. An illustration of the four primary immunological deficiencies associated with pancreatic cancer.
Figure 1. An illustration of the four primary immunological deficiencies associated with pancreatic cancer.
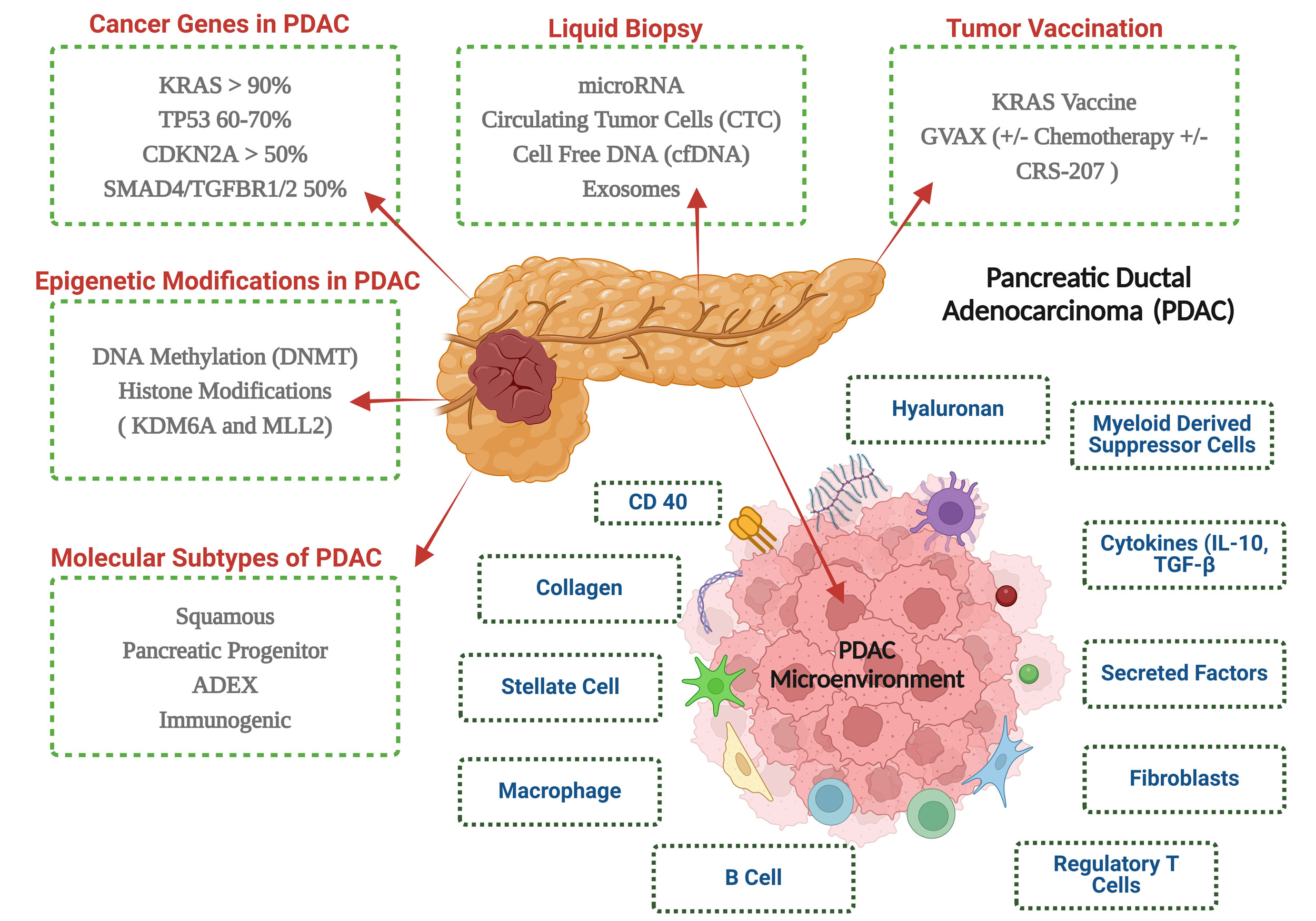 Figure 2. An overview of the key elements of pancreatic ductal adenocarcinoma molecular pathology and immunotherapy.
Figure 2. An overview of the key elements of pancreatic ductal adenocarcinoma molecular pathology and immunotherapy.
The tumor suppressor p53, which is mutated in 75% of pancreatic ductal adenocarcinomas (PDAC), stops working in advanced stages of the disease [21]. p53, sometimes referred to as the "guardian of the genome," typically causes cancer cells to undergo apoptosis. Mothers Against Decapentaplegic Homolog 4 (SMAD4) is another important mutation linked to the downregulation of SMAD4 protein production. Signaling from the TGF-beta family of ligands is mediated by SMAD4 and can either stimulate apoptosis or cellular growth. About 55% of pancreatic adenocarcinomas have SMAD4 mutations [22]. The progression of PC is also influenced by abnormal autocrine and paracrine signaling. Numerous mechanisms that promote invasion, and proliferation are stimulated by signaling molecules such insulin-like growth factor 1, fibroblast growth factor, and hepatocyte growth factor. The majority of clinical experiments that have attempted to target these pathways have failed.
By downregulating elements of the antigen presentation machinery, including MHC class I, PDAC tumors further avoid immune detection [29]. Alongside this, there is a rise in the expression of inhibitory immunological checkpoint ligands and apoptotic regulatory proteins which promote apoptotic resistance [30]. By stimulating cancer-associated fibroblasts (CAFs), PDAC physically separates anti-tumor immune cells from the TME [23]. By rearranging the extracellular matrix and depositing collagen, CAFs promote fibrosis and cause a desmoplastic reaction. This physical barrier caused by the desmoplastic stroma, which can make up 50-80% of the tumor volume, hinders vascularization, prevents anti-tumor immune cells from infiltrating, and reduces the effectiveness of systemic treatments [31]. Notably, this barrier is observed in both primary and metastatic pancreatic tumors [32]. Together, these factors contribute to the immunosuppressive nature of the PDAC TME, accounting for the poor response to both chemotherapy and immunotherapy [33].
Natural nonsulfated glycosaminoglycan hyaluronan is a key component of the EM. Although hyaluronan is found in normal connective, neural, and epithelial tissues, high amounts of it in cancers have been associated with a worse prognosis, faster tumor growth, and a lower chance of survival. Pegylated hyaluronidase (PEGPH20) was used in an attempt to break down this barrier because of the high interstitial pressures inside tumors that hinder perfusion. However, there was no increase in survival outcomes for the entire study group in a phase II trial that combined gemcitabine, nab-paclitaxel, and PEGPH20 [36]. Furthermore, unexpected toxicities resulted from hyaluronan's extensive presence. The FDA issued a clinical hold on the trial following the observation of unexpected arterial and venous thrombosis. This necessitated protocol modifications, including the mandatory use of lovenox anticoagulation to mitigate life-threatening blood clot risks.
Similarly, the SWOG trial that combined FOLFIRINOX and PEGPH20 initially relied on aspirin for prophylaxis. This approach was found to be insufficient, prompting amendments to require lovenox instead. However, the trial was ultimately halted by the data and safety monitoring committee due to a lack of clinical efficacy.
The first SMO antagonist to be discovered was cyclopamine, which had notable efficacy in preclinical studies [41]. Cyclopamine therapy in PC cell lines led to an elevation of E-cadherin and a downregulation of snail. In orthotopic models, cyclopamine efficiently decreased metastasis and shrunk primary tumors when paired with gemcitabine. In a phase II trial, gemcitabine and vismodegib, a second-generation SMO antagonist authorized for advanced basal cell carcinoma, were assessed together. The main results of this double-blind trial was progression-free survival (PFS), and 106 patients were randomly assigned. The median PFS for the combination treatment was 4 months, while the median PFS for gemcitabine alone was 2.5 months. At 6.9 and 6.1 months, respectively, the overall survival rates did not differ appreciably [42].
Unexpectedly, Saridegib (IPI-926) demonstrated worse outcomes in a phase II randomized trial, where patients receiving the combination therapy had shorter survival [43]. It was hypothesized that stromal degradation facilitated metastasis. Interestingly, a phase I trial combining Saridegib with FOLFIRINOX showed promising results, reporting a 67% response rate. However, this trial was not continued, and the development of Saridegib was ultimately discontinued [44].
The Notch signaling pathway is highly conserved and plays a key part in regulating neurogenesis during embryonic development. While the precise molecular mechanisms by which Notch contributes to PC pathogenesis remain unclear, it is known to interact with other signaling pathways such as MEK/ERK, Hedgehog (Hh), and Wnt, among others [45]. Targeting PC by inhibiting γ-secretase, an enzyme that activates Notch, is a unique treatment strategy. By blocking the epithelial-mesenchymal transition (EMT) and reducing the number of pancreatic CSCs, γ-secretase inhibitors (GSIs) have shown promise in reducing the proliferation [46]. Because tumor cells overexpress the notch ligand delta-like ligand 4 (DLL4), notch signaling is activated in CSCs [47]. By focusing on CSCs, the DLL4 inhibitor demcizumab has demonstrated promise in overcoming chemotherapy resistance [48].
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway has also been implicated in cancer progression. This pathway transmits signals from tyrosine kinase receptors and plays a role in mediating inflammation in both tumor and host tissues. In tumors with active KRAS mutations, STAT3 is especially crucial for the development of PDAC. Capecitabine and rufolitinib, a JAK1/JAK2 inhibitor that has been licensed for the treatment of high-risk myelofibrosis (MF) and hydroxyurea-intolerant/refractory polycythemia vera (PV), were studied together for pancreatic cancer. However, a phase III trial was discontinued because interim analysis revealed no discernible improvement [49].
In mice models of pancreatic cancer, STAT3 inhibition has been shown to be effective in slowing tumor growth [50]. The first-in-class cancer "stemness" inhibitor napabucasin targets the spherogenesis of CSCs and STAT3-driven gene transcription. Napapucasin demonstrated encouraging outcomes in early clinical trials. Diarrhea, nausea, vomiting, and electrolyte abnormalities were the most frequent side effects in a phase Ib/II trial with 71 participants; no unexpected toxicities were reported. According to preliminary statistics, the median overall survival was over 10.4 months, while the median PFS was over 7.1 months [51].
An estimated 3.9-35% of pancreatic tumors have acquired somatic mutations [54, 55]. The phase III NOVA research, which demonstrated the effectiveness of the PARP-1/2 inhibitor niraparib in treating patients with platinum-sensitive relapsed ovarian cancer, has validated this idea in the context of ovarian cancer. When compared to a placebo, the highest improvement in PFS was shown in cancers with germline BRCA mutations and in non-mutated BRCA tumors displaying homologous recombination deficiency (HRD) [56]. The HRD score is a measure of genomic instability that indicates a tumor's lack of homologous recombination DNA repair. Responses to PARP inhibitors and platinum-based chemotherapy have both been predicted using this score. Patients with refractory metastatic PC are presently being randomly assigned to FOLFIRI with or without ABT888 in the phase II trial SWOG S1513. Even though the data and safety monitoring committee recently ended this experiment, additional analyses are being carried out to assess responses according to the HRD score [57].
However, when compared to the standard of care for patients with stage II-IV cancer undergoing palliative treatment, a meta-analysis of four randomized controlled trials assessing cetuximab, an EGFR inhibitor, in PDAC revealed no discernible clinical improvement [60]. As a result, antibody-based immunomodulation has gained more attention. A 0% response rate in PD-L1 positive cases indicates that these antibody-based immune checkpoint medications have previously had poor response rates in non-dMMR PDAC patients [61]. In order to overcome this problem, antibody treatments are frequently investigated in conjunction with chemotherapy in an effort to produce synergistic tumor damage. The goal of this combined method is to increase the possibility for therapeutic success by combining the cytotoxic effects of chemotherapy with improved immunological detection of malignant cells.
By blocking immune checkpoint pathways, these therapies can inadvertently cause the immune system to target normal cells, resulting in various autoinflammatory side effects. The most common of these include hepatitis, pneumonitis, and thyroid dysfunction [62]. Bispecific T-cell engagers are a novel approach being investigated in acute myeloid leukemia and prostate cancer to address these issues. By pushing T cells closer to the tumor cells for improved recognition, these molecules' binding sites for both T cells and tumor-specific antigens increase specificity [63]. The FDA approved the anti-CD3 x anti-delta-like ligand 3 (anti-DLL3) bispecific antibody tarlatamab for the treatment of platinum-refractory small cell lung cancer (SCLC), demonstrating the therapeutic promise of this strategy [64]. Bispecific antibodies have a bright future in the therapy of solid malignancies, according to these studies.
TAVs have the potential to provide systemic efficacy as a customized treatment, according to preliminary data [68]. TAV usage is now being assessed in a number of active clinical experiments against standard care regimens for a variety of solid tumors, especially in patients with advanced disease. The results that have been published thus far are generally optimistic, even though many of these studies are still in their early phases and there is a lack of data. For example, it has been demonstrated that patients with moderate-to advanced-stage colorectal cancer respond especially well to the use of an autologous tumor cell-Bacillus Calmette–Guérin (BCG) vaccine (OncoVax), which mixes irradiation tumor cells with the TICE strain of Mycobacterium BCG [69]. Furthermore, Gemogenovatucel-T, a TAV made of autologous tumor cells transfected with a plasmid containing the GM-CSF gene (a cytokine that boosts the immune system) and a bifunctional short hairpin construct that inhibits furin, has been shown in multiple studies to have promise in the treatment of ovarian cancer patients [70]. Additionally, mRNA-based TAVs are showing promise as a therapy option for treatment-resistant solid tumors [66], but cell-based TAVs have been the most widely used technique in clinical trials and practice.
When paired with adjuvant mFOLFIRINOX and anti-PD-L1 treatment, recent developments in mRNA vaccines for PDAC have demonstrated encouraging results, especially in patients with resectable tumors (Table 1; NCT04161755). In a research by Rojas et al., this combination was administered to a small cohort of 16 patients, and 8 of them developed strong, neoantigen-specific T-cell responses. At an 18-month follow-up, all 8 responders showed no disease progression, indicating a considerably longer PFS than non-responders [71]. The homogeneous character of the patient group-all participants had curable diseases and could be treated with surgery was a major study drawback. On the other hand, the metastatic form of PC at diagnosis frequently contributes to the disease's low survival statistics [72]. Therefore, more investigation is required to determine whether these neoantigen-targeting T cells can detect and eradicate cancer cells that are circulating in the circulation and whether they can target metastatic lesions. Enhancing therapy options for PC patients may require applying this strategy to a larger population. All things considered, these results point to the great potential of therapeutic antitumor vaccinations (TAVs) as a PDAC treatment approach.
Chimeric antigen receptor T-cell (CAR-T) therapy involves extracting a patient's T lymphocytes, genetically modifying them to recognize and destroy cancer cells [77]. CAR-T cells are highly beneficial due to their ability to provide both immediate and long-lasting effects, as they continue to proliferate through clonal expansion [78, 79]. Five generations of CAR-T cells have developed throughout time to increase their proliferative potential, decrease toxicity, and improve targeting accuracy. B-cell cancers can now be treated using second-generation CAR-T cells. Compared to the first generation, these cells have an extra costimulatory molecule that improves T-cell responses and persistence. An extra costimulatory domain speeds up tumor clearance even more in the third generation. The fourth generation, referred to as "armored CAR-T cells" or TRUCKs (T-cells Redirected for antigen-Unrestricted Cytokine-initiated Killing), may be useful in solid tumors because it uses an interleukin inducer to get past immunosuppressive components in TME [80]. The fifth generation, still in development, adds a signal transducer and activator of transcription 3 (STAT3) binding site to promote engagement with the JAK-STAT pathway, thereby enhancing immune stimulation [81]. Efforts to improve CAR-T cell therapies for PDAC and other solid tumors are extensive and focus on enhancing efficacy.
The goal of research on localized delivery strategies, like those used for glioblastoma, is to minimize systemic toxicity while more accurately delivering CAR-T cells to the tumor site. Furthermore, by enlisting local immune cells, armored CAR-T cells that produce cytokines have been demonstrated to fight the immunosuppressive TME. This strategy may increase efficacy even more when combined with other immunotherapies [82]. In a range of solid cancers, including gastrointestinal cancers research into "tandem CAR-T cells" that contain two different antibody fragments has also shown promise in enhancing the detection and elimination of tumors with heterogeneous antigen expression [83]. These developments show how CAR-T cell therapies are continuously being improved to meet the particular difficulties associated with treating solid tumors.
None.
Availability of data and materials
Data and materials are available on request from the authors.
Ethical policy
Not applicable.
Author contributions
AFN contributed to draft, critical revision of the article and figure production; MA provided the idea and submitted the final version online.
Competing interests
I declare that there is no conflict of interest regarding the publication of this document.
Funding
None.
- Siegel RL, Miller KD, Jemal A: Cancer statistics, 2018. CA Cancer J Clin 2018, 68(1): 7-30.
- Cabasag CJ, Arnold M, Rutherford M, Bardot A, Ferlay J, Morgan E, Little A, De P, Dixon E, Woods RR et al: Pancreatic cancer survival by stage and age in seven high-income countries (ICBP SURVMARK-2): A population-based study. Br J Cancer 2022, 126(12): 1774-1782.
- Mahadevan KK, LeBleu VS, Ramirez EV, Chen Y, Li B, Sockwell AM, Gagea M, Sugimoto H, Sthanam LK, Tampe D et al: Elimination of oncogenic KRAS in genetic mouse models eradicates pancreatic cancer by inducing FAS-dependent apoptosis by CD8+ T cells. Dev Cell 2023, 58(17): 1562-1577.
- Pan M, Jiang C, Zhang Z, Achacoso N, Alexeeff S, Solorzano AV, Tse P, Chung E, Sundaresan T, Suga JM et al: TP53 gain-of-function and non–gain-of-function mutations are associated with differential prognosis in advanced pancreatic ductal adenocarcinoma. JCO Precis Oncol 2023, 7: e2200570.
- Ho WJ, Jaffee EM, Zheng L: The tumour microenvironment in pancreatic cancer-clinical challenges and opportunities. Nat Rev Clin Oncol 2020, 17(9): 527-540.
- Lennon AM, Wolfgang CL, Canto MI, Klein AP, Herman JM, Goggins M, Fishman EK, Kamel I, Weiss MJ, Diaz LA et al: The early detection of pancreatic cancer: what will it take to diagnose and treat curable pancreatic neoplasia?. Cancer Res 2014, 74(13): 3381-3389.
- Foster DS, Jones RE, Ransom RC, Longaker MT, Norton JA: The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3(18): e99911.
- Dvorak HF: Tumors: wounds that do not heal-redux. Cancer Immunol Res 2015, 3(1): 1-11.
- Ramezani F, Panahi Meymandi AR, Akbari B, Tamtaji OR, Mirzaei H, Brown CE, Mirzaei HR: Outsmarting trogocytosis to boost CAR NK/T cell therapy. Mol Cancer 2023, 22(1): 183.
- Garfall AL, Cohen AD, Susanibar-Adaniya SP, Hwang WT, Vogl DT, Waxman AJ, Lacey SF, Gonzalez VE, Fraietta JA, Gupta M et al: Anti-BCMA/CD19 CAR T cells with early immunomodulatory maintenance for multiple myeloma responding to initial or later-line therapy. Blood Cancer Discov 2023, 4(2): 118-133.
- Farhangnia P, Mollazadeh Ghomi S, Mollazadehghomi S, Delbandi AA: Current clinical landscape of immunotherapeutic approaches in pancreatic cancer treatment. InGastrointestinal Cancers: An Interdisciplinary Approach 2023, 29 (pp. 327-380). Cham: Springer Nature Switzerland.
- Zheng L, Ding D, Edil BH, Judkins C, Durham JN, Thomas DL, Bever KM, Mo G, Solt SE, Hoare JA et al: Vaccine-induced intratumoral lymphoid aggregates correlate with survival following treatment with a neoadjuvant and adjuvant vaccine in patients with resectable pancreatic adenocarcinoma. Clin Cancer Res 2021, 27(5): 1278-1286.
- Riquelme E, Maitra A, McAllister F: Immunotherapy for pancreatic cancer: more than just a gut feeling. Cancer Discov 2018, 8(4): 386-388.
- Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo W, Zhao F, You L, Zheng L, Zhang T et al: Targeting hypoxic tumor microenvironment in pancreatic cancer. J Hematol Oncol 2021, 14(1): 14.
- Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G: Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol 2020, (12): 725-741.
- Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Löhr JM, Friess H: Pancreatic cancer microenvironment. Int J Cancer 2007, 121(4): 699-705.
- Hong SM, Vincent A, Kanda M, Leclerc J, Omura N, Borges M, Klein AP, Canto MI, Hruban RH, Goggins M et al: Genome-wide somatic copy number alterations in low-grade PanINs and IPMNs from individuals with a family history of pancreatic cancer. Clin Cancer Res 2012, 18(16): 4303-4312.
- Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W et al: Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997, 57(15): 3126-3130.
- Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW et al: The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell 1998, 92(6): 713-723.
- Weinberg RA: The retinoblastoma protein and cell cycle control. Cell 1995, 81(3): 323-330.
- Mangray S, King TC: Molecular pathobiology of pancreatic adenocarcinoma. Front Biosci 1998, 3: D1148-1160.
- Hahn SA, Schutte M, Shamsul Hoque AT, Moskaluk CA, Da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH et al: DPC4, a candidate tumor suppressor gene at human chromosome 18q21. 1. Science 1996, 271(5247): 350-353.
- Karamitopoulou E: Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br J Cancer 2019, 121(1): 5-14.
- Das M, Zhou X, Liu Y, Das A, Vincent BG, Li J, Liu R, Huang L: Tumor neoantigen heterogeneity impacts bystander immune inhibition of pancreatic cancer growth. Trans Oncol 2020, 13(12): 100856.
- Gong R, Ren H: Targeting chemokines/chemokine receptors: A promising strategy for enhancing the immunotherapy of pancreatic ductal adenocarcinoma. Signal Transduct Target Ther 2020, 5(1): 149.
- Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton G: Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother 2011, 60(10): 1419-1430.
- Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, Beatty GL, Furth EE, Vonderheide RH: Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med 2020, 217(8): e20190673.
- Yamamoto T, Yanagimoto H, Satoi S, Toyokawa H, Yamao J, Kim S, Terakawa N, Takahashi K, Kwon AH: Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J Surg Res 2012, 173(2): 299-308.
- Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn AS, Mukhopadhyay S, Lin EY, Parker SJ et al: Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581(7806): 100-105.
- Bowers JS, Bailey SR, Rubinstein MP, Paulos CM, Camp ER: Genomics meets immunity in pancreatic cancer: Current research and future directions for pancreatic adenocarcinoma immunotherapy. Oncol Rev 2019, 13(2): 430.
- Haqq J, Howells LM, Garcea G, Metcalfe MS, Steward WP, Dennison AR: Pancreatic stellate cells and pancreas cancer: current perspectives and future strategies. Eur J Cancer 2014, 50(15): 2570-2582.
- Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H et al: Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res 2015, 21(15): 3561-3568.
- Schizas D, Charalampakis N, Kole C, Economopoulou P, Koustas E, Gkotsis E, Ziogas D, Psyrri A, Karamouzis MV: Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat Rev 2020, 86: 102016.
- Kota J, Hancock J, Kwon J, Korc M: Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett 2017, 391: 38-49.
- Erez N, Truitt M, Olson P, Hanahan D: Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010, 17(2): 135-147.
- Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, Braiteh F, Ritch PS, Zalupski MM, Bahary N et al: HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol 2018, 36(4): 359-366.
- Klonisch T, Wiechec E, Hombach-Klonisch S, Ande SR, Wesselborg S, Schulze-Osthoff K, Los M: Cancer stem cell markers in common cancers–therapeutic implications. Trends Mol Med 2008, 14(10): 450-460.
- Ingham PW, McMahon AP: Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001, 15(23): 3059-3087.
- Hahn H, Wicking C, Zaphiropoulos PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S et al: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85(6): 841-851.
- Takebe N, Harris PJ, Warren RQ, Ivy SP: Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011, 8(2): 97-106.
- Incardona JP, Gaffield W, Kapur RP, Roelink H: The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125(18): 3553-3562.
- Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L et al: Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J clin Oncol 2015, 33(36): 4284-4292.
- Sarvepalli D, Rashid MU, Rahman AU, Ullah W, Hussain I, Hasan B, Jehanzeb S, Khan AK, Jain AG, Khetpal N et al: Gemcitabine: a review of chemoresistance in pancreatic cancer. Critical Reviews™ in Oncogenesis 2019, 24(2): 199-212.
- Ko AH, LoConte NK, Kantoff E, Ross RW, Trehu EG, Tempero MA, Kelley RK, Venook AP, Wu T, Kindler HL et al: A phase Ib trial of FOLFIRINOX plus saridegib, an oral hedgehog (Hh) inhibitor, in pts with advanced pancreatic cancer (PDAC). J Clin Oncol 2012, 30: 3105
- Lin NY, Distler A, Beyer C, Philipi-Schöbinger A, Breda S, Dees C, Stock M, Tomcik M, Niemeier A, Dell'Accio F et al: Inhibition of Notch1 promotes hedgehog signalling in a HES1-dependent manner in chondrocytes and exacerbates experimental osteoarthritis. Ann Rheum Dis 2016, 75(11): 2037-2044.
- Palagani V, El Khatib M, Kossatz U, Bozko P, Müller MR, Manns MP, Krech T, Malek NP, Plentz RR: Epithelial mesenchymal transition and pancreatic tumor initiating CD44+/EpCAM+ cells are inhibited by γ-secretase inhibitor IX. PLoS One 2012, 7(10): e46514.
- Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, Matsui W, Eberhart CG, Maitra A, Feldmann G et al: Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res 2009, 15(7): 2291-2301.
- Gracian AC, Dean A, Muñoz A, Hidalgo M, Pazo-Cid R, Martin M, Mercade TM, Lipton L, Harris M, Manzano-Mozo JL et al: YOSEMITE: A 3 arm double-blind randomized phase 2 study of gemcitabine, paclitaxel protein-bound particles for injectable suspension, and placebo (GAP) versus gemcitabine, paclitaxel protein-bound particles for injectable suspension and either 1 or 2 truncated courses of demcizumab (GAD). Ann Oncol 2017, 28: v211.
- Hurwitz H, Van Cutsem E, Bendell JC, Hidalgo M, Li CP, Garrido M, Macarulla TM, Sahai V, Sama AR, Greeno E et al: Two randomized, placebo-controlled phase 3 studies of ruxolitinib (Rux)+ capecitabine (C) in patients (pts) with advanced/metastatic pancreatic cancer (mPC) after failure/intolerance of first-line chemotherapy: JANUS 1 (J1) and JANUS 2 (J2). Am Soc Clin Oncol 2017.
- Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, Leggett D, Li W, Pardee AB, Li CJ et al: Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci 2015, 112(6): 1839-1844.
- Bekaii-Saab TS, Starodub A, El-Rayes BF, O'Neil BH, Shahda S, Ciombor KK, Noonan AM, Hanna WT, Sehdev A, Shaib WL et al: A phase Ib/II study of cancer stemness inhibitor napabucasin (BBI-608) in combination with gemcitabine (gem) and nab-paclitaxel (nabPTX) in metastatic pancreatic adenocarcinoma (mPDAC) patients (pts). Am Soc Clin Oncol 2017.
- Murphy KM, Brune KA, Griffin C, Sollenberger JE, Petersen GM, Bansal R, Hruban RH, Kern SE: Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res 2002, 62(13): 3789-3793.
- O'Reilly EM, Lowery MA, Segal MF, Smith SC, Moore MJ, Kindler HL, Golan T, Segal A, Salo-Mullen EE, Hollywood E et al: Phase IB trial of cisplatin (C), gemcitabine (G), and veliparib (V) in patients with known or potential BRCA or PALB2-mutated pancreas adenocarcinoma (PC). J Clin Oncol 2014, 32, 4023.
- Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, Mead S, Jones MD, Samra JS, Gill AJ et al: Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res 2015, 21(9): 2029-2037.
- Turner N, Tutt A, Ashworth A: Hallmarks of'BRCAness' in sporadic cancers. Nat Rev Cancer 2004, 4(10): 814-819.
- Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I et al Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 2016, 375(22): 2154-2164.
- Chiorean EG, McDonough S, Philip PA, Swisher EM, Pishvaian MJ, Guthrie K, Lowy AM, Hochster HS: Randomized phase II study of 2nd-line FOLFIRI versus modified FOLFIRI with PARP inhibitor ABT-888 (veliparib)(NSC-737664) in metastatic pancreatic cancer (mPC): SWOG S1513. Clin Cancer Res 2021, 27(23): 6314-6322.
- Goydel RS, Rader C: Antibody-based cancer therapy. Oncogene 2021, 40(21): 3655-3664.
- Li Z, Wang M, Yao X, Luo W, Qu Y, Yu D, Li X, Fang J, Huang C: Development of a novel EGFR-targeting antibody-drug conjugate for pancreatic cancer therapy. Target Oncol 2019, 14(1): 93-105.
- Forster T, Huettner FJ, Springfeld C, Loehr M, Kalkum E, Hackbusch M, Hackert T, Diener MK, Probst P: Cetuximab in pancreatic cancer therapy: a systematic review and meta-analysis. Oncology 2020, 98(1): 53-60.
- Ott PA, Bang YJ, Piha-Paul SA, Razak AR, Bennouna J, Soria JC, Rugo HS, Cohen RB, O’Neil BH, Mehnert JM et al: T-cell-nflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol 2019, 37(4): 318-327.
- Kwok G, Yau TC, Chiu JW, Tse E: Kwong YLl. Pembrolizumab (Keytruda). Hum Vaccin Immunother 2016, 12(11): 2777-2789.
- Palecki J, Bhasin A, Bernstein A, Mille PJ, Tester WJ, Kelly WK, Zarrabi KK: T-Cell redirecting bispecific antibodies: a review of a novel class of immuno-oncology for advanced prostate cancer. Cancer Biol Ther 2024, 25(1): 2356820.
- FDA U: FDA grants accelerated approval to tarlatamab-dlle for extensive stage small cell lung cancer [Internet]. 2024.
- Tsai HJ: Clinical cancer chemoprevention: From the hepatitis B virus (HBV) vaccine to the human papillomavirus (HPV) vaccine. Taiwan J Obstet Gynecol 2015, 54(2): 112-115.
- Wang B, Pei J, Xu S, Liu J, Yu J: Recent advances in mRNA cancer vaccines: meeting challenges and embracing opportunities. Front Immunol 2023, 14: 1246682.
- Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C: Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther 2023, 8(1): 450.
- Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X: Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol 2022, 15(1): 28.
- Hanna, Jr MG: Immunotherapy with autologous tumor cell vaccines for treatment of occult disease in early stage colon cancer. Hum Vaccin Immunother 2012, 8(8): 1156-1160.
- Watkins DE, Craig DJ, Vellani SD, Hegazi A, Fredrickson KJ, Walter A, Stanbery L, Nemunaitis J: Advances in targeted therapy for the treatment of cervical cancer. J Clin Med 2023, 12(18): 5992.
- Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, Lihm J, Ceglia N, Guasp P, Chu A et al: Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618(7963): 144-150.
- Li Y, Feng A, Zheng S, Chen C, Lyu J: Recent estimates and predictions of 5-year survival in patients with gastric cancer: a model-based period analysis. Cancer Control 2022, 29: 10732748221099227.
- El-Kadiry AE, Rafei M, Shammaa R: Cell therapy: types, regulation, and clinical benefits. Front Med 2021, 8: 756029.
- Titov A, Zmievskaya E, Ganeeva I, Valiullina A, Petukhov A, Rakhmatullina A, Miftakhova R, Fainshtein M, Rizvanov A, Bulatov E et al: Adoptive immunotherapy beyond CAR T-cells. Cancers 2021, 13(4): 743.
- Okur FV, Brenner MK: Cellular immunotherapy of cancer. Immunotherapy of Cancer: Methods Mol Biol 2010, 651: 319-345.
- Hegazi A, Rager LE, Watkins DE, Su KH: Advancing Immunotherapy in Pancreatic Cancer. Intl Mol Sci 2024, 25(21): 11560.
- Sterner RC, Sterner RM: CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 2021, 11(4): 69.
- Sadelain M, Brentjens R, Rivière I: The basic principles of chimeric antigen receptor design. Cancer Discov 2013, 3(4): 388-398.
- Yeo D, Giardina C, Saxena P, Rasko JE: The next wave of cellular immunotherapies in pancreatic cancer. Mol Ther Oncolytics 2022, 24: 561-576.
- Tomasik J, Jasiński M, Basak GW: Next generations of CAR-T cells-new therapeutic opportunities in hematology?. Front Immunol 2022, 13: 1034707.
- Mehrabadi AZ, Ranjbar R, Farzanehpour M, Shahriary A, Dorostkar R, Hamidinejad MA, Ghaleh HE: Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed Pharmacother 2022, 146: 112512.
- Tang L, Pan S, Wei X, Xu X, Wei Q: Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol Ther 2023, 31(11): 3146-3162.
- Yang M, Tang X, Zhang Z, Gu L, Wei H, Zhao S, Zhong K, Mu M, Huang C, Jiang C et al: Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10(17): 7622-7634.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.