
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Adverse effects of the cancer therapy on osteoclast-mediated bone loss in patients with cancers: a challenge
Manh Tien Tran11Department of Molecular Medicine, Institute of Biotechnology, UT Health San Antonio, TX 78229, USA.
Correspondence: Manh Tien Tran (Department of Molecular Medicine, Institute of Biotechnology, UT Health San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229; E-mail: trantienmanh1508@gmail.com).
Asia-Pacific Journal of Oncology 2022, 3: 10-15. https://doi.org/10.32948/ajo.2022.12.29
Received: 28 Dec 2022 | Accepted: 29 Dec 2022 | Published online: 31 Dec 2022 
Key words chemotherapy drugs, osteoclastogenesis, bone loss, osteoporosis
Bone remodeling is stringently regulated by osteoblasts (OBs) and osteoclasts (OCs), which are responsible for bone formation and bone resorption, respectively. OC progenitors, derived from monocyte/macrophage lineage, are differentiated into mature, multinucleated OCs via a series of osteoclastogenic signaling pathways [3, 4]. In principle, OC differentiation and maturation, also known as osteoclastogenesis, are primarily induced by the binding of two critical cytokines, including (1) the receptor of nuclear factor kappa-B ligand (RANKL) to its specific RANK receptor and (2) the macrophage colony-stimulating factor (M-CSF) to c-fms receptor in cell surface of OC progenitors [5, 6]. RANKL/RANK signaling triggers the commitment of monocyte/macrophage precursors to the OC lineage and subsequently mature, multinucleated OCs via predominantly activating six downstream signaling cascades comprising (1) the nuclear factor of activated T cells cytoplasmic-1 (NFATc-1); (2) nuclear factor kappa B (NF-κB); (3) phosphatidylinositol 3-kinase (PI3K/Akt); (4) Jun N-terminal kinase (JNK); (5) extracellular signal-regulated kinase (Erk); and (6) p38 mitogen-activated protein kinase (MAPK) [7-9]. Nevertheless, the precise molecular mechanisms underlying the activation of these signaling pathways remains disputable. In addition to RANKL/RANK signaling axis itself, accumulating evidence illuminates that interferon (IFN) γ, IFN-β and immunoreceptor tyrosine-based activation motif (ITAM) play the specific roles in regulating osteoclastogenesis by cross-talking with one or more RANKL/RANK signaling pathways [10]. In bone, OBs and bone marrow stromal cells serve as primary soures of RANKL, M-CSF and osteoprotegerin (OPG). OPG functions as a decoy for RANKL, therefore inhibiting the binding of RANKL to RANK receptor, thereby alleviating osteoclastogenesis [11, 12]. Indeed, in vivo studies substantiated that mice deficient for the gene encoding either RANKL or RANK repcetor developed osteopetrosis (a genetically etiological state of elevated bone mass) owing to the impaired OC formation [13]. Moreover, OPG-deficient mice developed early onset of osteoporosis [14] whereas transgenic mice overexpressing OPG exhibited osteopetrosis in mice [15]. In this review, I have struggled to summarize the published papers revealing the effects of cancer therapies on regulating bone cell- induced bone loss in the patients with cancers.
Bone remodeling is tightly regulated by specialized cells including OBs, OCs and osteocytes. Oestrogen plays an important role in maintaining bone mass in adult women by suppressing bone remodeling and maintaining a balance between osteoblast and osteoclastic activities. Oestrogen alleviates the osteoblastic production of resorptive cytokines, including RANKL, M-CSF and tumor necrosis factor, and at the same time increases the production of antireceptive cytokines (mainly osteoprotegerin) [11]. This leads to increased osteoclastic apoptosis and increased osteoblast activity. However, when the oestrogen levels are deficient, there is an increase in remodeling imbalance, resulting in osteoclastic activity, subsequently leading to deeper resorption spaces [19]. There is also some evidence that the ability of the OBs to refill these spaces may be impaired. Moreover, the deeper resorption spaces result in perforation of trabecular plates and loss of architectural elements, further weakening the skeleton in regions such as the vertebrae and distal forearm which contain large amounts of cancellous bone. These abnormalities in postmenopausal women appear to be more severe in women with postmenopausal osteoporosis [20]. Furthermore, the intestinal absorption and the renal re-absorption of calcium are also diminished. This triggers a rise in the serum-parathyroid hormone that activates OCs, thereby ruling in an increase in a net bone loss. Besides, a recent study has reported that aromatase inhibitor (exemestane) had a stimulatory effect on osteoblastic acitivity in human OB-like cells expressing aromatase genes [21]. On the contrary, whether aromatase inhibitors regulate osteoclastic activity in vitro is completely unknown.
In ovariectomized rats and mice, it was found that the aromatase inhibitors increased bone mass, and bone strength [22]. However, clinical data showed that aromatase inhibitors increased bone resorption and decreased BMD at the hip and lumbar spine, suggesting that it is likely these inhibitors potentially increase the risk of bone fractures. More crucially, from these data, it is suggested that there are remarkable differences in bone metabolism between rats and human. Tamoxifen, also named nolvadex or soltamox, a selective estrogen receptor modulator (SERM), used to trat all stages of hormone receptor-positive breast cancer in women and men, had a positive effect on BMD, but not likely to decline the risk of bone fractures [23]. Nevertheless, aromatase inhibitors (letrozole, anastrozole and exemestane) had negative effects on BMD and enhanced the risk of bone fractures in the patients with breast cancers. Interestingly, previous studies have shown some drugs such as cathepsin K antagonists and RANKL antibodies (denusomab), might be of potential interest in aromatase inhibitor-induced osteoporosis in the patients with the post-menopausal osteoporosis.
ADT is considered to be the primary or adjuvant treatment for non-metastatic prostate cancer [28]. Owing to decificiency of adrogens and estrogens that are crucial for bone remodeling and maintenance, the skeleton is virtually compromised in the male patients with prostate cancer during ADT. In fact, the ADT-treated patients sustained variable degrees of bone loss with an enhanced risk of fragility fractures [29]. Mechanistically, it was because of a remodeling imbalance between osteoclastic and osteoblastic activities wherein OCs resorbed the deeper resorption bone spaces. Besides, it was feasible that OBs were incapable of refilling these OC-resorbed bone spaces. It was reported that male patients undergoing ADT are four time more likely to develop considerable bone loss [30]. A previous study of prostate cancer patients who survived more than 5 years after diagnosis of prostate cancer, 19.4% of those receiving ADT had a higher tendency to develop the fragility fracture [30]. Consequently, bone health management in the patients with prostate cancer receiving ADT is urgently required to control bone loss-associated fractures. The antiresorptive drugs that weakened osteoclastic bone destruction were tested in controlled randomized patient groups receiving ADT. For example, the oral bisphosphonates, pyrophosphate analogs (alendronate and risedronate) and intravenous bisphosphonates (pamidronate and zoledronic acid) suppress bone turnover via interfering with the internal enzymatic system and disrupting the cytoskeleton of OCs, thereby triggering OC apoptosis, stabilizing bone mass and reducing the risk of fragility fracture in the osteoporosis patients, in general, and the patients with prostate cancer receiving ADT, in particular [31]. Accordingly, bisphosphonates have become the primary therapy for managing skeletal conditions characterized by increased OC-mediated bone resorption.
Methotrexate (MTX)
A commonly used anti-metabolite, causes a significant reduction of BMD [32]. In chemotherapeutic memchanism, MTX inhibits RNA/DNA synthesis via weakening the dihydrofolate reductase. After MTX treatment, bone formation is attenuated, and bone loss is elevated. These outcomes have been associated with several specificfactors such as, for example, (1) abolished circulating levels of vitamin D3, (2) depressed of OB precursor pool within bone marrow, and (3) varied response of bone cells towards mechanical loading [33]. Though these studies indicated BMD is impaired as a major result of a derease in total bone synthesis, increasing evidence, however, it was suggested that the impaired BMD was also cause by enhanced OC-mediated bone resorption. Indeed, it was reported that an increase in the density of OC precursors and an elevation of OC maturation in bone marrow in rats after short-term MTX treatment [34]. Furthermore, prolonged administration of MTX at a low dose resulted in osteopenia, linked to enhanced OC activity and recruitment [35]. Together, these studies indicate that the major reason leading to bone loss after MTX chemotherapy originates from increased OC differentiation, maturation and bone-resorbing activity.
Doxorubicin (DOX)
An anthracycline drug originally isolated from the fungus Streptomyces peucetius. This drug has been clinically used to treat a variety of cancers, comprising breast cancer, bladder cancer, lymphoma, and acute lymphocytic leukemia. DOX has been reported to inhibit topoisomerase II and induce cell death by generating reactive oxygen species (ROS) [36]. In spite of its role as a chemotherapeutic agent, emerging proof has demonstrated its side effects on bone physiology. For instance, DOX-treated breast cancer patients exhibited low bone mineral density, and especially DOX-treated children underwent a long-term bone damage [37]. At the cellular level, DOX was reported to diminish cell division and differentiation derived from MC3T3 mouse [38]. Also, DOX was found to reduce trabecular bone volume and cortical bone thickness in rabbits, mice and rats. DOX treatment promoted the activation of TGFβ/Smad signaling pathways, which played a critical role in accelerating OC maturation, as well as suppressing OB differentiation [37]. Additionally, TGFβ may also regulate the physiological characteristics of bone via modulating bone mass and bone milieu. Indeed, other studies have demonstrated that blocking excess TGFβ in bone milieu, either by the specific antibodies or by small molecule inhibitors, promoted bone formation. Ultimately, increase in TGFβ level was associated with the generation of reactive oxidative species (ROS), which is also consistent with the fact that DOX increases oxidative stress.
1,3-Dibenzyl-5-fluorouracil (5-FU)
It and its oral produgs including S1 and capecitabine are the important components of most chemotherapeutic regimens of which efficiencies was reported in the treatment of the patients with various neoplasms such as, for instance, head and neck squamous cell carcinoma (SCC), gastrointestinal SCC and adenocarcinoma (ADC), breast cancer, stomach cancer, colon cancer, etc. 5-FU inhibits thymidylate synthase, an enzyme required for the synthesis of thymine nucleotide, an important component of DNA and RNA [39]. Mechanistically, 5-FU and other chemotherapeutic agents including cisplatin, epotoside, cyclophosphamide have been shown to cause severe osteopenia by promoting OC differentiation as well as suppressing OB formation [40]. Nonetheness, it was the fact that the underlying molecular mechanisms of chemotherapy-induced bone loss still remain largely unknown. Moreover, no safe and cose-effective treatments against chemotherapy-induced bone loss are available. As abovementioned, only the antiresorpive therapies using bisphosphonates are well documented to abolish bone resorption, and remarkably elevate bone mass, and therefore possess some potential efficacies in reducing osteoporosis.
Though the mechanisms underlying the augmentation of OC-induced bone loss in the patients with cancers during the period of chemotherapeutic treatments is little known (Figure 1), it has a severe impact on bone metabolism and architecture.
Altogether, it seems to be unavoidable that cancer therapies lead to OC-induced bone defects in the patients with cancers. It is, however, urgently required to develop the special programs of drug development that is conjugated with chemotherapeutic agents to treat cancer patients to alleviate OC-induced bone resorption and/or enhance OB-induced bone formation (Table 1), thereby maintaining bone metabolism at the physiological level in these patients. The followings are several drug conjugates, even limited, which possess the potential impact on abolishing OC-induced bone loss in the cancer patients during cancer therapies.
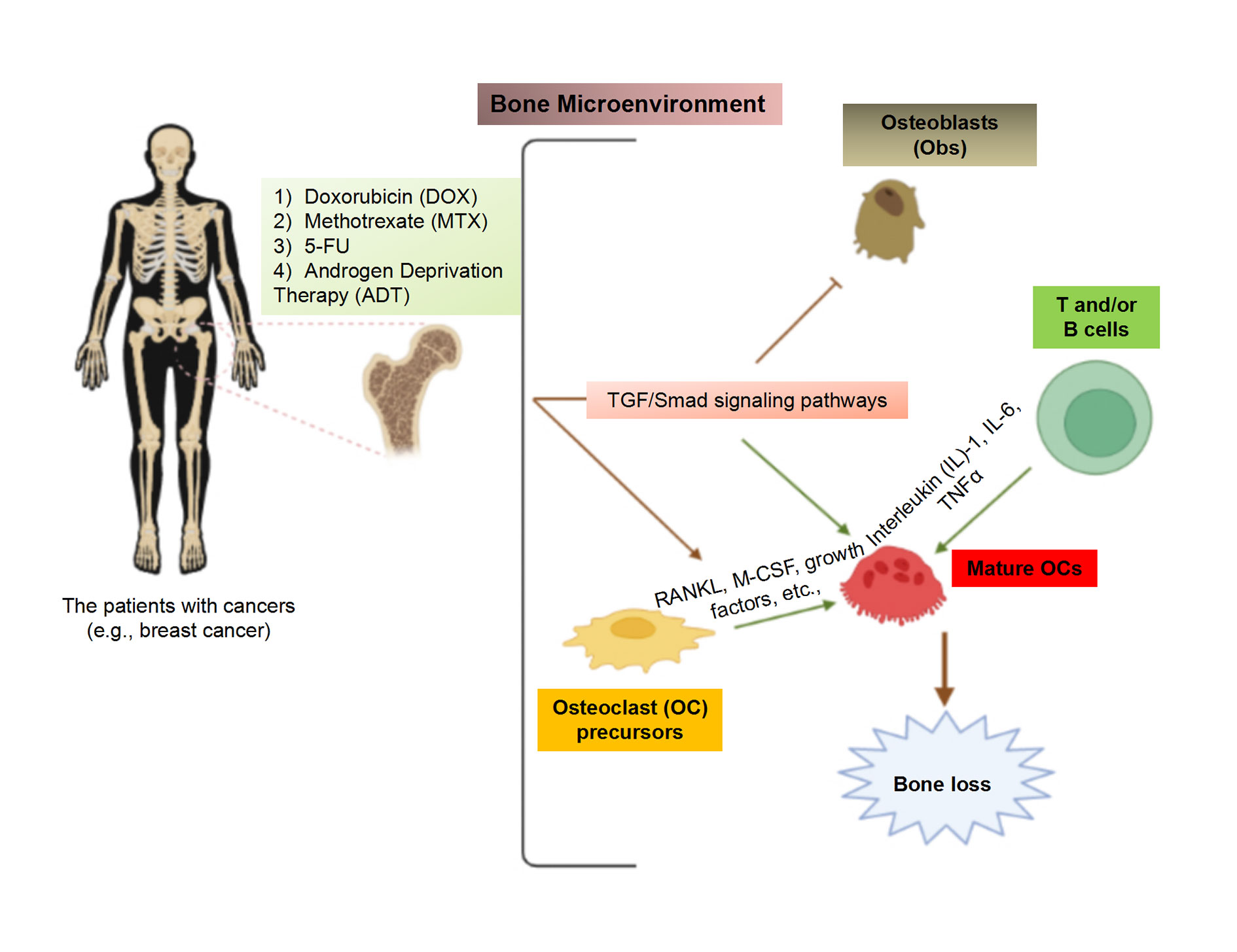 Figure 1. Cancer therapies causes the OC-induced bone loss in the patients with cancers. The chemotherapeutic agents such as DOX, MTX and 5-FU, and ADT commonly trigger the activation of TGF/Smad signaling pathway, which is crucial for stimulating osteoclastogensis. Besides, the levels of RANKL and M-CSF are elevated, accelerating OC differentiation and maturation. Besides, these chemotherapeutic drugs alo promoted the production of T cell-secreted cytokines such as IL1, IL6 and TNFα, which lead to osteoclastogenesis, thereby triggering bone loss in the patients with cancer.
Figure 1. Cancer therapies causes the OC-induced bone loss in the patients with cancers. The chemotherapeutic agents such as DOX, MTX and 5-FU, and ADT commonly trigger the activation of TGF/Smad signaling pathway, which is crucial for stimulating osteoclastogensis. Besides, the levels of RANKL and M-CSF are elevated, accelerating OC differentiation and maturation. Besides, these chemotherapeutic drugs alo promoted the production of T cell-secreted cytokines such as IL1, IL6 and TNFα, which lead to osteoclastogenesis, thereby triggering bone loss in the patients with cancer.
|
Table 1. The summary of the drug conjugates currently used in combination with cancer theparies to treat the patients with cancers. |
||
|
Cancer Therapies |
Types of cancers |
Drug conjugates |
|
Aromatase |
Breast cancer |
Aromatase inhibitors (letrozole, anastrozole and exemestane) [22] |
|
Androgen Deprivation Therapy (ADT) |
Prostate cancer |
Oral bisphosphonates, pyrophosphate analogs (alendronate and risedronate), intravenous bisphosphonates (pamidronate and zoledronic acid) [31] |
|
Methotrexate (MTX) |
Leukemia, osteosarcomas, bladder cancer, breast cancer, etc. |
Alendrolate (ALN), zoledronic acid (ZA) are third-generation bisphosphonates [41] |
|
Doxorubicin (DOX) |
Leukemia, bone sarcoma, breast cancer, gastric cancer, head and neck cancer, liver cancer, etc. |
Anti-TGFβ antibody [37] , resveratrol [42], MitoTEMPO [42] |
|
1,3-Dibenzyl-5-fluorouracil (5-FU) |
Breast cancer, head and neck cancer, colon cancer, skin cancer, etc. |
Emu oil (EO) [40], Complement nutritionals (calcium and vitamin D), [43], bisphosphonate zoledronic acid (ZA) [44, 45], aromatase inhibitors [46] |
Not applicable.
Author contributions
The author contributed solely to the work.
Competing interests
The author declares that there are no conflicts of interest.
Funding
Not applicable.
- LeBoff MS, Greenspan SL, Insogna KL, Lewiecki EM, Saag KG, Singer AJ, Siris ES: The clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int 2022, 33(10): 2049-2102.
- Michaud LB, Buzdar AU: Risks and benefits of aromatase inhibitors in postmenopausal breast cancer. Drug Saf 1999, 21(4): 297-309.
- Väänänen HK, Zhao H, Mulari M, Halleen JM: The cell biology of osteoclast function. J Cell Sci 2000, 113 ( Pt 3): 377-381.
- Kim JH, Kim N: Signaling Pathways in Osteoclast Differentiation. Chonnam Med J 2016, 52(1): 12-17.
- Feng Y, Tran MT, Lu Y, Htike K, Okusha Y, Sogawa C, Eguchi T, Kadowaki T, Sakai E, Tsukuba T, et al: Rab34 plays a critical role as a bidirectional regulator of osteoclastogenesis. Cell Biochem Funct 2022, 40(3): 263-277.
- Okusha Y, Tran MT, Itagaki M, Sogawa C, Eguchi T, Okui T, Kadowaki T, Sakai E, Tsukuba T, Okamoto K: Rab11A Functions as a Negative Regulator of Osteoclastogenesis through Dictating Lysosome-Induced Proteolysis of c-fms and RANK Surface Receptors. Cells 2020, 9(11): 2384.
- Tran MT, Okusha Y, Feng Y, Morimatsu M, Wei P, Sogawa C, Eguchi T, Kadowaki T, Sakai E, Okamura H, et al: The Inhibitory Role of Rab11b in Osteoclastogenesis through Triggering Lysosome-Induced Degradation of c-Fms and RANK Surface Receptors. Int J Mol Sci 2020, 21(24): 9352.
- Tran MT, Okusha Y, Feng Y, Sogawa C, Eguchi T, Kadowaki T, Sakai E, Tsukuba T, Okamoto K: A novel role of HSP90 in regulating osteoclastogenesis by abrogating Rab11b-driven transport. Biochim Biophys Acta Mol Cell Res 2021, 1868(10): 119096.
- Tran MT, Okusha Y, Htike K, Sogawa C, Eguchi T, Kadowaki T, Sakai E, Tsukuba T, Okamoto K: HSP90 drives the Rab11a-mediated vesicular transport of the cell surface receptors in osteoclasts. Cell Biochem Funct 2022, 40(8): 838-855.
- Feng X: RANKing intracellular signaling in osteoclasts. IUBMB Life 2005, 57(6): 389-395.
- Tran MT: Overview of Ca(2+) signaling in lung cancer progression and metastatic lung cancer with bone metastasis. Explor Target Antitumor Ther 2021, 2(3): 249-265.
- Tran MT: Roles of renin-angiotensin system in the regulation of prostate cancer bone metastasis: a critical review. Ann Urol Oncol 2021, 4(1): 31-41.
- Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, et al: RANK is essential for osteoclast and lymph node development. Genes Dev 1999, 13(18): 2412-2424.
- Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al: osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998, 12(9): 1260-1268.
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al: Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997, 89(2): 309-319.
- Rago V, Romeo F, Aquila S, Montanaro D, Andò S, Carpino A: Cytochrome P450 aromatase expression in human seminoma. Reprod Biol Endocrinol 2005, 3: 72.
- Bhardwaj P, Au CC, Benito-Martin A, Ladumor H, Oshchepkova S, Moges R, Brown KA: Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J Steroid Biochem Mol Biol 2019, 189: 161-170.
- Hyder T, Marino CC, Ahmad S, Nasrazadani A, Brufsky AM: Aromatase Inhibitor-Associated Musculoskeletal Syndrome: Understanding Mechanisms and Management. Front Endocrinol (Lausanne) 2021, 12: 713700.
- Weitzmann MN, Pacifici R: Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest 2006, 116(5): 1186-1194.
- Tella SH, Gallagher JC: Prevention and treatment of postmenopausal osteoporosis. J Steroid Biochem Mol Biol 2014, 142: 155-170.
- Miki Y, Suzuki T, Hatori M, Igarashi K, Aisaki KI, Kanno J, Nakamura Y, Uzuki M, Sawai T, Sasano H: Effects of aromatase inhibitors on human osteoblast and osteoblast-like cells: a possible androgenic bone protective effects induced by exemestane. Bone 2007, 40(4): 876-887.
- Lelovas PP, Xanthos TT, Thoma SE, Lyritis GP, Dontas IA: The laboratory rat as an animal model for osteoporosis research. Comp Med 2008, 58(5): 424-430.
- Lee SJ, Cha CD, Hong H, Choi YY, Chung MS: Adverse effects of tamoxifen treatment on bone mineral density in premenopausal patients with breast cancer: a systematic review and meta-analysis. Breast Cancer 2024, 31(4): 717-725.
- Khalid AB, Krum SA: Estrogen receptors alpha and beta in bone. Bone 2016, 87: 130-135.
- Shevde NK, Bendixen AC, Dienger KM, Pike JW: Estrogens suppress RANK ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-Jun repression. Proc Natl Acad Sci U S A 2000, 97(14): 7829-7834.
- Robinson LJ, Yaroslavskiy BB, Griswold RD, Zadorozny EV, Guo L, Tourkova IL, Blair HC: Estrogen inhibits RANKL-stimulated osteoclastic differentiation of human monocytes through estrogen and RANKL-regulated interaction of estrogen receptor-alpha with BCAR1 and Traf6. Exp Cell Res 2009, 315(7): 1287-1301.
- Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O'Brien CA: RANKL (Receptor Activator of NFκB Ligand) Produced by Osteocytes Is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J Biol Chem 2016, 291(48): 24838-24850.
- Liede A, Hallett DC, Hope K, Graham A, Arellano J, Shahinian VB: International survey of androgen deprivation therapy (ADT) for non-metastatic prostate cancer in 19 countries. ESMO Open 2016, 1(2): e000040.
- Cianferotti L, Bertoldo F, Carini M, Kanis JA, Lapini A, Longo N, Martorana G, Mirone V, Reginster JY, Rizzoli R, et al: The prevention of fragility fractures in patients with non-metastatic prostate cancer: a position statement by the international osteoporosis foundation. Oncotarget 2017, 8(43): 75646-75663.
- Bienz M, Saad F: Androgen-deprivation therapy and bone loss in prostate cancer patients: a clinical review. Bonekey Rep 2015, 4: 716.
- Jung J, Park JS, Righesso L, Pabst AM, Al-Nawas B, Kwon YD, Walter C: Effects of an oral bisphosphonate and three intravenous bisphosphonates on several cell types in vitro. Clin Oral Investig 2018, 22(7): 2527-2534.
- Fan C, Georgiou KR, King TJ, Xian CJ: Methotrexate toxicity in growing long bones of young rats: a model for studying cancer chemotherapy-induced bone growth defects in children. J Biomed Biotechnol 2011, 2011: 903097.
- Smith EA, Frankenburg EP, Goldstein SA, Koshizuka K, Elstner E, Said J, Kubota T, Uskokovic M, Koeffler HP: Effects of long-term administration of vitamin D3 analogs to mice. J Endocrinol 2000, 165(1): 163-172.
- Liu Y, Cui Y, Chen Y, Gao X, Su Y, Cui L: Effects of dexamethasone, celecoxib, and methotrexate on the histology and metabolism of bone tissue in healthy Sprague Dawley rats. Clin Interv Aging 2015, 10: 1245-1253.
- May KP, West SG, McDermott MT, Huffer WE: The effect of low-dose methotrexate on bone metabolism and histomorphometry in rats. Arthritis Rheum 1994, 37(2): 201-206.
- Kalet BT, McBryde MB, Espinosa JM, Koch TH: Doxazolidine induction of apoptosis by a topoisomerase II independent mechanism. J Med Chem 2007, 50(18): 4493-4500.
- Rana T, Chakrabarti A, Freeman M, Biswas S: Doxorubicin-mediated bone loss in breast cancer bone metastases is driven by an interplay between oxidative stress and induction of TGFβ. PLoS One 2013, 8(10): e78043.
- Udagawa N, Takahashi N, Akatsu T, Sasaki T, Yamaguchi A, Kodama H, Martin TJ, Suda T: The bone marrow-derived stromal cell lines MC3T3-G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen cells. Endocrinology 1989, 125(4): 1805-1813.
- Zhang N, Yin Y, Xu SJ, Chen WS: 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules 2008, 13(8): 1551-1569.
- Raghu Nadhanan R, Abimosleh SM, Su YW, Scherer MA, Howarth GS, Xian CJ: Dietary emu oil supplementation suppresses 5-fluorouracil chemotherapy-induced inflammation, osteoclast formation, and bone loss. Am J Physiol Endocrinol Metab 2012, 302(11): E1440-1449.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.