
 Submit Manuscript
Submit ManuscriptNiclosamide exerts anti-tumor activity through generation of reactive oxygen species and by suppression of Wnt/ β-catenin signaling axis in HGC-27, MKN-74 human gastric cancer cells
Manish Kumar Jeengar1,5,*, Shravan Kumar2,*, Shweta Shrivastava2, Syamprasad N P1, Vladimir L. Katanaev3,4, Srinivas Uppugunduri5, V.G.M. Naidu1*
1Department of Pharmacology & Toxicology, National Institute of Pharmaceutical Education and Research (NIPER)-Guwahati, Mirza, Assam - 781125, India.
2Department of Pharmacology & Toxicology, National Institute of Pharmaceutical Education & Research (NIPER), Balanagar, Hyderabad, Telangana, 500037, India.
3Department of Pharmacology and Toxicology, Faculty of Biology and Medicine, University of Lausanne, Rue du Bugnon 27, Lausanne 1005, Switzerland.
4School of Biomedicine, Far Eastern Federal University, 8, Sukhanova str., Vladivostok, Russian Federation.
5Regional Cancer Center South East Sweden and Department of Clinical and Experimental Medicine, Linköping University, Linköping, Sweden.
Correspondence: Dr. V.G.M. Naidu ( Department of Pharmacology & Toxicology C/O NETES Institute of Technology & Science, NH-37, Shantipur, Parli Part, Mirza, Assam - 781125; E-mail: naiduvgm23@gmail.com, vgmnaidu@gmail.com).
*These authors contributed equally to this work.
Asia-Pacific Journal of Oncology 2020, 1: 18-30. https://doi.org/10.32948/ajo.2020.08.06
Received: 29 Dec 2019 | Accepted: 22 Jun 2020 | Published Online: 12 Aug 2020
Introduction Gastric carcinoma (GC) remains a therapeutic challenge despite having many potent drugs to treat. Various studies emphasized the role of dysregulated Wnt/β-catenin pathway in cancer. In the present study, we examined the anti-cancer effect of Niclosamide and its effect on the dysregulated β-catenin pathway in human gastric carcinoma cell lines.
Methods Cytotoxicity of compound to gastric cancer cell line was assessed by MTT cell viability assay, cell cycle analysis, and apoptosis assay was done using standard kits of Muse™ Cell Analyser. Reactive oxygen species (ROS) generation and mitochondrial membrane potential were analyzed by 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA) and tetraethylbenzimidazolylcarbocyanine iodide (JC-1) staining respectively. Protein expression studies were carried out by standard western blotting protocols.
Results Niclosamide treatment resulted in a dose-dependent inhibition of viability of the gastric carcinoma cell-lines induced cell cycle arrest in the G0/G1 phase and strongly induced apoptosis in a concentration-dependent manner by downregulating Cyclin-D1 and CDK4 levels, critical proteins required for G1-S phase progression. DCFDA and JC-1 staining results indicated that Niclosamide enhanced intracellular ROS generation and disrupted mitochondrial membrane potential. Furthermore, niclosamide treatment decreased the expression of NF-KB, Bcl-2 and increased the expression of Bax protein. Niclosamide treatment significantly decreased the β-catenin mediated transcriptional activity and down-regulated β-catenin levels and its downstream proteins cyclinD1, CDK-4, and c-myc expression and also impeded Akt phosphorylation, a common internode in the Wnt and Akt/mTOR signaling in HGC-27 cells.
Conclusion This study demonstrated that Niclosamide might become a promising therapeutic agent for the management of gastric cancer and further warrants its clinical trials in gastric cancer patients.
Key words Niclosamide exerts, reactive oxygen species, anti-tumor activity, MEM, NF-kB, PARP
Gastric cancer is the third most common cause of cancer- related death and ranks fifth in incidence worldwide, with about 70% of cases occurring in underdeveloped and developing countries. The etiology of gastric cancer is complex, including both dietary and non-dietary; over 80% of cases are attributed to Helicobacter pylori infection. Gastric cancer usually presents at more advanced invasive stages as a majority of patients remain asymptomatic in the earlier stages of malignancy [1]. The mainstays of treatment include surgical resection, peri-operative chemotherapy, and radiation in appropriate cases [2]. Despite recent progress in its detection and treatment strategies, gastric cancer has an abysmal prognosis with a median overall survival (OS) below one year in advanced metastatic patients [3]. There is thus a pressing need to develop effective chemotherapeutic agents for the management of gastric cancer.
Wnt/ β-Catenin signaling pathway is an evolutionarily conserved pathway that regulates crucial events such as cellular proliferation and apoptosis, multiple developmental aspects during embryogenesis, motility, organ development and is also implicated in tumorigenesis. The aberrant canonical Wnt/β-catenin signaling pathway plays a crucial role in the development of gastric carcinoma [4]. Mutations in multiple components of this pathway, mainly β-Catenin, have reported to be present in a major proportion of gastric cancers [2]. The intracellular β-catenin level is maintained by phosphorylation-dependent proteasomal degradation by the APC complex, which is the critical regulator of this canonical signaling pathway. Enhanced Wnt signaling leads to the deactivation of GSK-3β, a critical enzyme in the APC complex; this, in turn, leads to the accumulation of non-phosphorylated β-catenin, which enters the nucleus and causes transcription of genes critical for cell cycle progression and oncogenesis such as cyclin-D, CDK4, c-myc [5]. Pharmacological suppression of Wnt signaling has proved to be effective in the treatment of several cancers.
Drug repositioning of existing drugs for novel indications is an attractive strategy as it maximizes the value of an existing drug by reducing costs and timelines involved in the development of new drugs. In recent years, repositioned drugs accounted for almost 30 % of Food & Drug Administration (FDA) newly approved drugs and vaccines [6, 7].
Niclosamide (trade name: Niclocide), a salicylanilide derivative, also has anti-tumor activity apart from its well established anthelmintic activity. It has shown to exert its anti-tumor action by modulating multiple signaling pathways, including mTORC1, STAT3, NF-κB, and Wnt/ β-catenin signaling in a variety of cancers including breast, prostate cancers, and multiple myeloma [8, 9 10, 11]. Furthermore Niclosamide displayed strong inhibitory effect on the growth of cancer stem cells by modulating the stem cell critical pathways such as Wnt/ β-catenin, Notch, Hedgehog [12]. In this study, for the first time, we demonstrated the anti-tumor activity of Niclosamide on gastric cancer. We investigated the underlying molecular mechanism and its role inhibiting Wnt/ β-catenin, NF-κB, Akt signaling pathways in the gastric tumor cells.
Niclosamide was purchased from Santa Cruz Biotechnology, Inc. (Texas, U.S.A.). The powder was dissolved in dimethyl sulfoxide (DMSO) and stored as aliquots (20 mM) at -80 ºC. Roswell Park Memorial Institute-1640 (RPMI-1640) media, Minimal essential medium (MEM), Fetal bovine serum, Penicillin, Streptomycin, Trypsin/EDTA were purchased from Invitrogen Life Technologies, USA. Acridine orange (AO), 4′,6-diamidino-2-phenylindole (DAPI), Acrylamide, Ammonium acetate, 2’,7’–dichlorofluorescein diacetate (DCFDA), Dithiotheritol, Dihydrogen sodium phosphate, EGTA, tetraethylbenzimidazolylcarbocyanine iodide (JC-1), Methylthiazolyldiphenyl-tetrazolium bromide (MTT), Nonidet P-40, N1, N1-Dimethyl bis acrylamide, NADH, Propidium Iodide, Proteinase K, RNAse A, Sodium pyrophosphate, Tween 20, Triton-X-100 and all other reagents unless mentioned were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Primary antibodies included poly (ADP-ribose) polymerase (PARP) (116/89 kDa), NF-κB (P65) (65 kDa), Bcl-2 (28 kDa), Bax (20 kDa), Cytochrome C (14 kDa), p53 (53 kDa), total Akt (60 kDa), Ser 473 phosphorylated Akt (p-Akt) (60 kDa), p-4E-BP1 (21 kDa), p-70S6K1 (70 kDa) and Actin (42 kDa). Horseradish peroxidase-conjugated secondary anti-mouse and anti-rabbit antibodies were purchased from Cell Signaling Technology, Danvers, MA, USA.
Cell lines
Gastric cancer cell lines used: HGC-27 (Gastric cancer undifferentiated, mucin-producing- stomach, lymph node meta-epithelial-like) and MKN-74 (Moderately differentiated adenocarcinoma. Cell growth is slow - stomach, liver meta - epithelial-like) procured from Ricken cell bank, Japan. HGC-27 was cultured in MEM medium, and MKN-74 cell lines were cultured in RPMI supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and100 µg/mL streptomycin. The cells were maintained in a humidified incubator at 37o with 5% CO2 and 95% air. Cell lines were sub-cultured by digesting enzymatically with 0.25 % trypsin/1 mM EDTA solution when they reached approximately 70–80 % confluence.
Cell viability studies
The effects of Niclosamide on cell viability of HGC-27 and MKN-74 cell lines were assessed by MTT assay [13]. Cells were plated in flat-bottomed, 96-well microtiter plates at a density of 5 × 103 cells per well in 100µl medium and were allowed to attach for overnight in a CO2 incubator. After 12-24 hrs, cells were treated with different concentrations of Niclosamide and standard Wnt inhibitor Salinomycin (100 μM, 25 μM, 6.25 μM, 1.56 μM, 0.39 μM, and 0 μM). After 48 hrs of treatment, cells were treated with 100µl of MTT reagent dissolved in a medium (0.5 mg/ml) and kept for 4 hrs at 37°C. The formazan crystals were solubilized with 200 µl of DMSO. The percentage of cell viability is directly proportional to the absorbance of the formazan product, which was measured at 570 nm using a Multimode plate reader (SpectraMax M4, USA).
Morphological observation by AO/EB staining and DAPI staining
Annexin V and Propidium Iodide (PI) staining assay
Annexin V and Propidium Iodide (PI) staining assay was carried out using the method given by Kloesch et al., with minor modifications [15]. Briefly, to confirm the apoptosis induced by Niclosamide on HGC-27 and MKN-74, cells in a six-well plate were treated with 0.5 µM,1 µM, 2 µM and 2.5 µM, 5 µM, 10 µM concentrations of Niclosamide in HGC-27 and MKN-74 cell lines respectively for 48hrs. Then cells were treated with MuseTM Annexin-V & Dead Cell reagent (Merck-Millipore, Germany) as per instructions provided by the manufacturer and analyzed by Muse™ Cell Analyzer (Merck-Millipore, Germany).
Cell cycle analysis
Cell cycle analysis was performed using the method given by UV Mallavadhani et al., with slight modifications [16]. For this assay, HGC-27 and MKN-74 cells were seeded in 6-well plates at a density of 1 × 105 cells/ml for 24 hrs. After incubation, cells were treated with Niclosamide at 0.5 µM, 1 µM and 2 µM in HGC-27 cells. Then the cells were collected, washed, and fixed in 70% ethanol in PBS at -20oC. After overnight incubation, fixed cells were pelleted and stained with Propidium iodide in the presence of RNase A containing Triton X-100 for 30 min at 37oC in the dark and about 5000 events were analyzed in a Muse™ Cell Analyzer (Merck-Millipore, Germany).
Detection of intracellular ROS by DCFDA method
Intracellular ROS determination was assessed by the DCFDA method as given by Kim HJ et al. with minor modifications. The HGC-27 and MKN-74 cells were seeded into a 96- well black plate and cultured for 24hrs. After treatment with Niclosamide 1 µM, 2 µM, and 5 µM, 10 µM concentrations both HGC-27 and MKN-74 cell lines respectively for 1 hrs in presence and absence of N-acetyl-L-cysteine (NAC) (3 mM), the cells were incubated with 10 µM DCF-DA at 37 ºC for 15 min. The nonfluorescent DCFDA oxidized by intracellular ROS and converted into highly fluorescent DCF. The fluorescent intensity of DCF was monitored at excitation (498 nm) and emission (530 nm) wavelengths using blue filter by fluorescence microscope (Nikon 80i, Japan) at a magnification of 200X [17].
JC-1 staining
The HGC-27 and MKN-74 cells were seeded into a 96-well black plate and cultured for 24 hrs. After 24 hrs, attached cells were treated with Niclosamide at concentrations of 0.5 µM,1 µM, 2 µM and 2.5 µM, 5 µM, 10 µM in HGC-27 and MKN-74 cell lines respectively for 48 hrs. Then after 48 hrs, cells were washed with PBS, and JC-1 dye is added to the cells and incubated for 30 min. The images were visualized using a blue filter under a fluorescent microscope at an excitation wavelength of 450 nm and emission at 490 nm [18].
Scratch wound healing assay
HGC-27 Cells were grown in MEM supplemented with 10% FBS. Cells were seeded into 6-well tissue culture plate, and after 24 hrs of growth, they reached ~70-80% confluence as a monolayer. The monolayer was scratched gently and slowly with a new 1 ml pipette tip across the center of the well. After scratching, well was washed twice with medium to remove the detached cells. Wells were replenished with fresh medium & treated with different concentrations of Niclosamide. Cells were grown for an additional 24 hrs. Cells were then washed twice with 1x PBS and fixed with formalin for 30 minutes, and a microscope took monolayer image at a magnification of 200X [19].
Cell adhesion assay
Cell adhesion assay was carried out by a colorimetric method based on staining cells with the dye crystal violet [20]. Briefly, a 24-well culture plate was coated with 10 μg/mL of type I collagen for 24 hrs at 4 °C, washed twice with PBS, and then plates were blocked with 1% BSA in MEM for 1 hr. Cells treated with various concentrations of Niclosamide in MEM containing 10% FCS were plated at a density of 1 × 105 cells per coated well, followed by 1 hrs incubation at 37 °C. After washing unattached cells with PBS, adherent cells were fixed in ethanol, stained with 0.1% crystal violet, and then photographed under microscopy.
Western blotting
The effect of Niclosamide on protein expression of various downstream proteins of the Wnt signaling pathway was studied by using Western blotting technique [21]. After treatment with different concentrations of Niclosamide for 24 hrs in HGC-27 cell line, cells were collected and washed twice with 1X PBS, then lysed in Radioimmunoprecipitation assay (RIPA) buffer with Protease cocktail mixture & phosphatase inhibitor under gentle rotation for 20 min at 40C, and the supernatant was collected by centrifugation at 12,500g for 20 min at 40C and stored at -200C or used immediately. Protein concentration was determined by the BioRad Protein detection reagent (BioRad). Equal amounts of protein were separated on SDS polyacrylamide gels. The separated proteins were blotted or transferred onto polyvinyl difluoride (PVDF) membrane (PolyScreen, NEN Life Sciences, USA) and following blocking (5% nonfat dry milk in TBS-Tween (0.1%), and probed with relevant primary and HRP-conjugated secondary antibody. Membranes were visualized on Kodak BioMax MR-1 films, and further, the protein bands were subjected to a densitometry analysis using ImageJ software. The membranes were reprobed with β-actin antibodies as an internal control and to ensure equal loading. Each Western blot shown is representative of at least three independent experiments.
TopFlash Luciferase reporter assay
HTB19 (BT-20) triple-negative breast cancer cells (ATCC) were stably transfected with M50 Super 8x TOPFlash plasmid (Addgene, plasmid 12456) at the 20:1 ratio with pcDNA3.1 to provide antibiotic resistance. The best-responding clone was selected and expanded; this stable cell line is referred to as HTB19-Tf throughout the text. Luciferase reporter activity measurements were performed mostly as described [22] with several modifications. The HTB19-Tf cells were seeded in 100µl DMEM medium containing 10% FCS at ~10000 cells/well. After overnight attachment, the cells were additionally transfected by the pCMV-RL plasmid for constitutive expression of Renilla luciferase kindly provided by Konrad Basler [23] using X-tremeGENE 9 reagent (Roche) according to the manufacturer's protocol. After 24 hrs, cells were stimulated by 0.5µg/ml mouse Wnt3a purified as described [21] with or without drug compound for 12 hrs. The medium was subsequently removed, and 15µl of the lysis buffer (25mM Glycylglycine pH 7.8, 1% Triton X-100, 15mM MgSO4, 4mM EGTA, 1mM DTT) were added into each well. After incubation for 5 minutes at room temperature, the 96-well plate was analyzed in the Victor3 Multilabel Counter (PerkinElmer) with a two-channel dispensing unit primed with the buffer solutions for firefly and Renilla luciferase activity measurements prepared as described [24]. The final volumes dispensed per well were 50µl of firefly and 50µl of Renilla solutions.
Statistical analysis
All results were expressed as Mean ± SEM. The intergroup variation between various groups was assessed by one-way analysis of variance (ANOVA) using Graph Pad Prism, version 5.0, and the comparison between groups was conducted by "Bonferroni’s Multiple Comparison Test." Results were considered statistically significant when p < 0.05.
In order to investigate whether Niclosamide has a direct cytotoxic effect on gastric cancer cells, we tested the effect of Niclosamide on cell viability in two gastric cancer cell lines using the MTT assay, Niclosamide treatment for 48 hrs resulted in a dose-dependent decrease in cell viability. IC50 of Niclosamide was found to be 1.82 µM and 9.38 µM in HGC-27 and MKN-74 cell lines, respectively (Fig. 1A). Qualitative evaluation using phase contrast images also showed a dose-dependent decrease in the number of cells (Fig.1B & 1C). These results were compared with the standard Wnt inhibitor drug salinomycin IC50 was found to be 10µM and 35µM HGC-27 and MKN-74 cell lines, respectively.
Niclosamide induces apoptosis in gastric cancer cells
We performed a morphological assessment of Niclosamide treated cells using acridine orange/ethidium bromide staining and DAPI staining to test whether Niclosamide treatment causes apoptosis. Acridine orange/ ethidium bromide staining clearly showed that both cell lines exhibit the characteristic features of apoptosis-like chromatin condensation, membrane blubbing, and apoptotic body formation upon treatment with Niclosamide (Fig. 2A & 2B). DAPI staining revealed characteristic apoptotic features such as the horseshoe-shaped nucleus and nuclear fragmentation in Niclosamide treated cells when compared to control samples (Fig. 2C &2D).
We performed annexin V-FITC-Propidium Iodide staining of gastric cancer cells treated with Niclosamide for 24 hrs to confirm the apoptotic effects of Niclosamide further. Increased number of early apoptotic cells were observed after Niclosamide treatment for 0.5 µM (7.15%), 1 µM (27.60%) and 2 µM (39.65%) compared to the control (6.0%) in HGC-27 cells whereas 2.5 µM (8.65%), 5 µM (12.40%) and 10 µM (20.60%) compared to the control (8.16%) in MKN-74 cells (Fig. 3). These findings suggest that Niclosamide triggers apoptosis in gastric cancer cells in a dose-dependent manner.
Niclosamide arrests cell cycle at G0/G1 phase in gastric cancer cells
The cell cycle analysis by propidium iodide staining is a valuable method to determine the phase in which the cell cycle has been arrested by Niclosamide treatment. A dose-dependent increased percentage of G0/G1 cells was observed after 24 hrs treatment with Niclosamide (Fig. 4A & 4B). The G0/G1 population was only 41.3% in control HGC-27 cells, whereas the percentage of these cells increased to 42.5, 58.2, and 60.3 % after treatment with 0.5 µM, 1 µM, and 2 μM concentrations of Niclosamide respectively.
3.4 Niclosamide induces loss of mitochondrial membrane potential by increased ROS generation in gastric cancer cells
It is generally accepted that the loss of mitochondrial membrane potential invariably results in cell death due to the release of pro-apoptotic factors from mitochondria and a decrease in ATP production in mitochondria [16, 25]. To investigate the effect of Niclosamide on the mitochondrial membrane potential, we performed JC-1 staining. Niclosamide treated cells exhibited green color fluorescence in a dose-dependent manner compared to red color fluorescence exhibited by the control cells (Fig. 4E & 4F), indicating the loss of mitochondrial membrane potential.
Increased intracellular ROS generation often disrupts mitochondrial membrane potential and causes apoptosis by oxidation of mitochondrial membrane transition pore and release of cytochrome C [26, 27, 28]. Thus we next sought to determine the effect of Niclosamide on cellular ROS levels in gastric cancer cells using cell-permeant fluorogenic dye DCF-DA. Treatment with Niclosamide for 3 hrs caused a significant increase of ROS in these cancer cells at concentrations of 1, 2, 5, and 10 µM in HGC-27 and MKN-74 cells, respectively. Treatment with NAC reversed Niclosamide-induced ROS in the cancer cells (Fig. 4C & 4D). Overall, these results indicate that Niclosamide induces disruption of mitochondrial membrane potential by increasing intracellular ROS generation.
Niclosamide inhibits cell adhesion and migration
Cancer metastasis is an essential mediator of cancer-related mortality and morbidity. Cancer metastasis involves tumor cell migration, invasion, and adhesion to the extracellular matrix. We used the cell-matrix adhesion assay to test the effect of Niclosamide on cell adhesion to the extracellular matrix. Niclosamide treatment inhibited adhesion of HGC-27 cells to the matrix in a dose-dependent manner (Fig. 5A).
Therefore, in order to examine whether Niclosamide could inhibit gastric cancer cell migration, we performed a scratch wound healing assay on the HGC-27cell line. After 24 hrs exposure to Niclosamide, as shown in Fig. 5B, Niclosamide-treated groups showed reduced migration capability in a concentration-dependent manner. These results show that Niclosamide can suppress gastric cancer cell migration and adhesion in a concentration-dependent manner.
Niclosamide induces apoptosis through modulation of anti-apoptotic and pro-apoptotic factors by mitochondria mediated intrinsic apoptotic pathway
As demonstrated earlier, Niclosamide increases intracellular ROS generation and disrupts mitochondrial membrane potential in gastric cancer cells, which indicates the possible involvement of mitochondrial-mediated effects in the anti-tumor mechanisms of Niclosamide. Hence we further analyzed whether Niclosamide induced apoptosis occurs through the mitochondrial-mediated pathway by analyzing protein expression of pro-apoptotic protein (Bax) and anti-apoptotic protein (Bcl-2).
The levels of Bcl-2 and Bax were quantified in control and treated cell lysates with β-actin as control (Fig. 6A & 6B). Increased levels of Bax, decreased protein levels of Bcl-2 were observed in a dose-dependent manner in response to the Niclosamide treatment. The Bax-to-Bcl-2 ratios in cells treated with 1 and 2 μM Niclosamide were 1.97 and 10.86, respectively. A collapse of mitochondrial membrane potential is closely associated with the release of the pro-apoptotic protein cytochrome C from mitochondria. We also determined the protein levels of cytochrome C. The results from Figure 6A indicate the release of cytochrome C into the cytosol.
PARP-1, a 116-kDa protein, is one of the substrates for caspase-3 and is cleaved to PARP-1 89-kDa protein upon activation of caspases in the apoptotic process. The appearance of the cleaved form of PARP-1 in cells treated with 1 µM and 2 μM Niclosamide treatment for 24 h suggests that caspase-mediated apoptosis is in play (Fig. 6A).
Collectively these results strongly suggest that Niclosamide induced cytotoxicity is mediated through the modulation of pro-apoptotic and anti-apoptotic proteins, release of cytochrome C, and PARP-1 cleavage.
Niclosamide inhibits Wnt/ β-catenin signaling pathway in gastric cancer cells.
Overactivation of the Wnt/β-catenin pathway and mutations in β-catenin are frequently observed in the marked proportion of gastric cancer [29]. Hence we next set forth to test the effect of Niclosamide on the transcriptional activity of Wnt/β-catenin signaling and thereby inhibition of its downstream targets that are involved in cell proliferation. We determined β-catenin mediated transcriptional activity in Niclosamide treated and control cells by using the Top Flash Luciferase reporter assay. 10 μM concentration of Niclosamide resulted in more than 40% inhibition of transcriptional activity (Fig. 7A). In the next series of experiments, we treated the HGC-27 cell line with Wnt3a, a known Wnt protein to examine the effects of Niclosamide in the presence of activated Wnt pathway. Stimulation with Wnt3a enhanced proliferation in a concentration-dependent manner, and Wnt3a at a concentration of 200ng/ml for 24 hr, cell viability of HGC-27 was increased 41.2% compared to untreated cells, as shown in Fig. 7C. Treatment with Niclosamide at 1, 2 µM concentration considerably decreased proliferation of Wnt3a ligand stimulated HGC-27 cells (Fig. 7D).
Further, we determined the effect of Niclosamide on the protein levels of Wnt / β-catenin upstream proteins and downstream targets in HGC-27 cells by western blotting. Phosphorylated forms of GSK-3β and total β-catenin decreased significantly in the treated cells compared to untreated cells without altering total GSK-3β levels (Fig. 8A). In another experiment, we stimulated cells with Wnt3a and found that it enhanced the total β-catenin levels when compared to untreated cells. Under similar stimulated conditions, Niclosamide treatment markedly decreased the total β-catenin levels (Fig. 7B). This reduced level of β-catenin indicates that Niclosamide might enhance the phosphorylation levels of β-catenin, which in turn results in its proteasomal degradation making it unavailable for nuclear transcriptional effects. Therefore, further protein expression of downstream effectors of β-catenin mediated pathways that play an important role in cell proliferation and cell cycle progression were also determined. Cyclin-D1, C-myc, and CDK-4 were all down-regulated in a dose-dependent manner in Niclosamide treated HGC-27 cells (Fig. 8A).
Overall, these results clearly indicate that Niclosamide downregulates the Wnt/ β-catenin signaling in gastric cancer cells, possibly by downregulating the phosphorylation of GSK-3β and decreasing the accumulation of β-catenin.
Niclosamide inhibits Akt/GSK-3β/NF-κB pathway activation in HGC-27 cells
Akt signaling pathway is an important signal transduction pathway that plays a critical role in cell survival and apoptotic process. Possible activation of Akt, which further phosphorylates and deactivates GSK-3β that, in turn, results in the stabilization of β-catenin, has been implicated in the activation of Wnt signaling pathway [30, 31]. Hence we checked whether Niclosamide could modulate the phosphorylation status of Akt by performing western blotting. As seen in Fig. 9A & 9B, we did not find any alterations in the total protein expression of Akt in Niclosamide-treated HGC-27 cells, whereas phosphorylated Akt protein at Ser473 decreased significantly in a dose-dependent manner after treatment with Niclosamide compared to untreated cells.
Recent reports showed that the NF-κB activation is also regulated by the Akt signaling pathway [32, 33, 34]. NF-κB family nuclear transcription factors play a cardinal role in inflammation, immune responses, as well as in various stages of tumor initiation, progression, and resistance to chemotherapy. Niclosamide treatment significantly decreased phosphorylation of p65 NF-κB in HGC-27 cells without altering total p65 NF-κB (Fig. 9A & 9B), suggesting that Niclosamide might inhibit NF-κB signaling pathway through inhibition of the common internode Akt. Collectively, these results provide evidence for a potential cross-talk between Wnt, Akt, and NF-κB pathway during Niclosamide-induced cell growth inhibition and apoptosis in gastric cancer cells. These results also demonstrate that Niclosamide induces potential modulation of these multiple signaling pathways, including Wnt, Akt, and NF-κB in the gastric cancer cells.
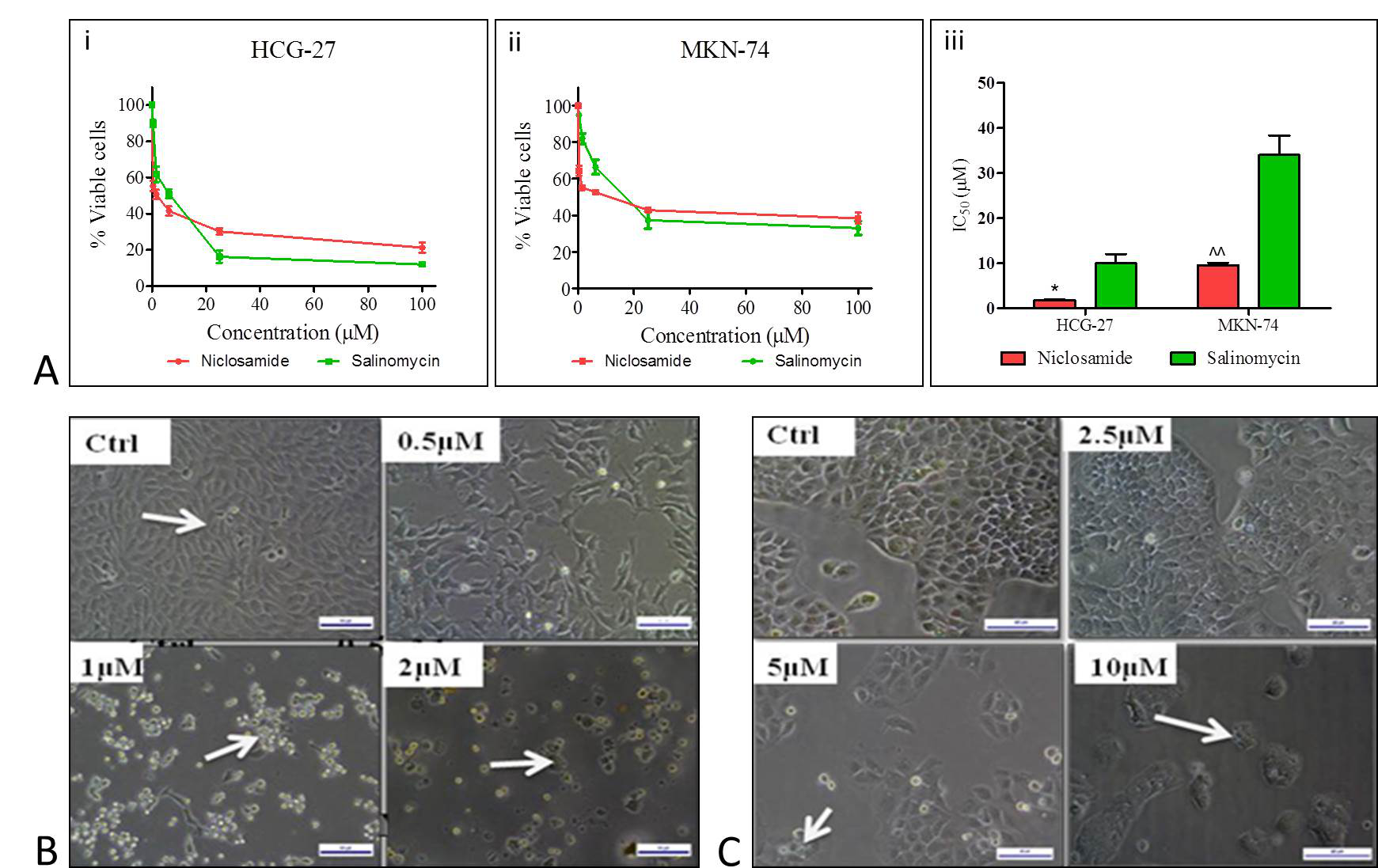 Figure 1. Shematic representation of anti-proliferation effect of Niclosamide on gastric cancer cells. (A) Dose response curves of niclosamide effect on viability of gastric cancer cells in i) HGC-27 ii) MKN-74 cells respectively as determined by MTT assay iii) IC-50 values of niclosamide in gastric cancer cell lines compared with standard drug salinomycin (B) Phase contrast images showing inhibition of proliferation in HGC-27 cells after treatment with 0, 0.5, 1,2 µM concentrations of Niclosamide (C) Phase contrast images showing change in the proliferation in MKN-74 cells after treatment with 0,2.5, 5, 10 µM concentrations of Niclosamide .
Figure 1. Shematic representation of anti-proliferation effect of Niclosamide on gastric cancer cells. (A) Dose response curves of niclosamide effect on viability of gastric cancer cells in i) HGC-27 ii) MKN-74 cells respectively as determined by MTT assay iii) IC-50 values of niclosamide in gastric cancer cell lines compared with standard drug salinomycin (B) Phase contrast images showing inhibition of proliferation in HGC-27 cells after treatment with 0, 0.5, 1,2 µM concentrations of Niclosamide (C) Phase contrast images showing change in the proliferation in MKN-74 cells after treatment with 0,2.5, 5, 10 µM concentrations of Niclosamide .
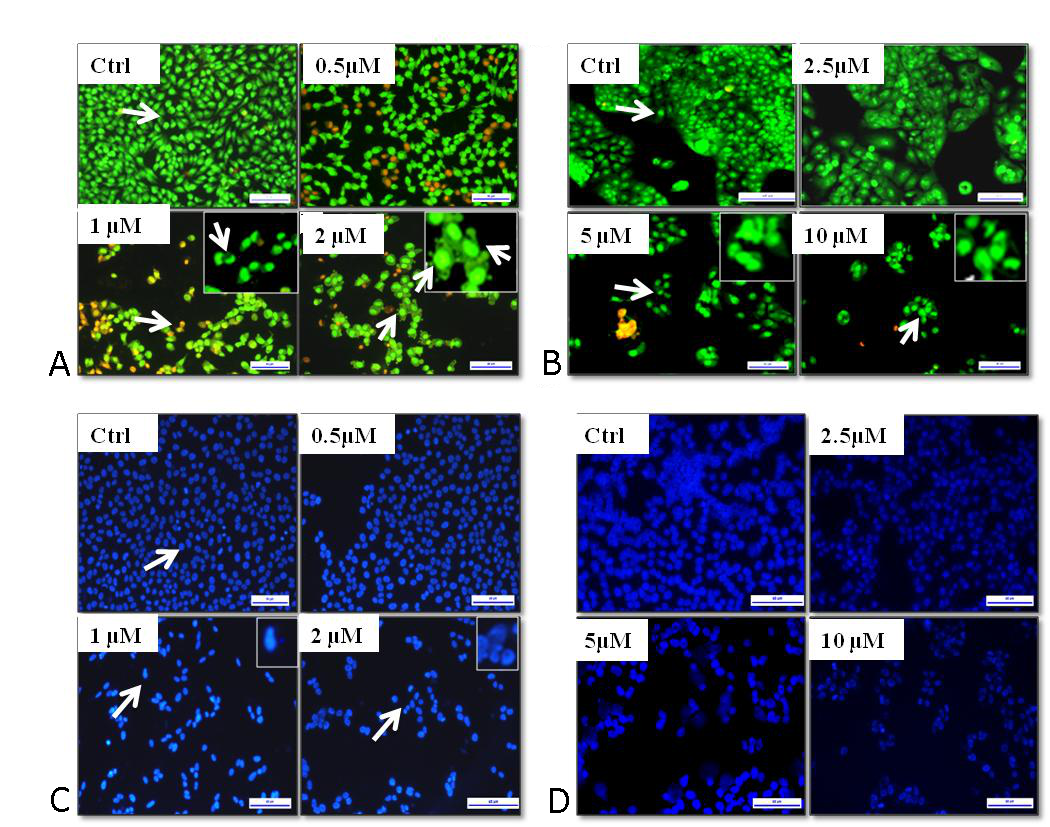 Figure 2. Effect of Niclosamide on morphological changes and apoptosis induction in gastric cancer cells, after treatment with 0 µM, 0.5 µM, 1 µM, and 2 µM in HGC-27 and 0 µM, 2.5 µM, 5 µM, and 10 µM in MKN-74. (A) & (B) Flourescent images obtained by AO/EB staining of Niclosamide treated cells with indicated concentrations for 48 h. in HGC-27, MKN-74 respectively (C) &(D) Nuclear morphology determined by DAPI staining in after 48h treatment in HGC-27, MKN-74 respectively .
Figure 2. Effect of Niclosamide on morphological changes and apoptosis induction in gastric cancer cells, after treatment with 0 µM, 0.5 µM, 1 µM, and 2 µM in HGC-27 and 0 µM, 2.5 µM, 5 µM, and 10 µM in MKN-74. (A) & (B) Flourescent images obtained by AO/EB staining of Niclosamide treated cells with indicated concentrations for 48 h. in HGC-27, MKN-74 respectively (C) &(D) Nuclear morphology determined by DAPI staining in after 48h treatment in HGC-27, MKN-74 respectively .
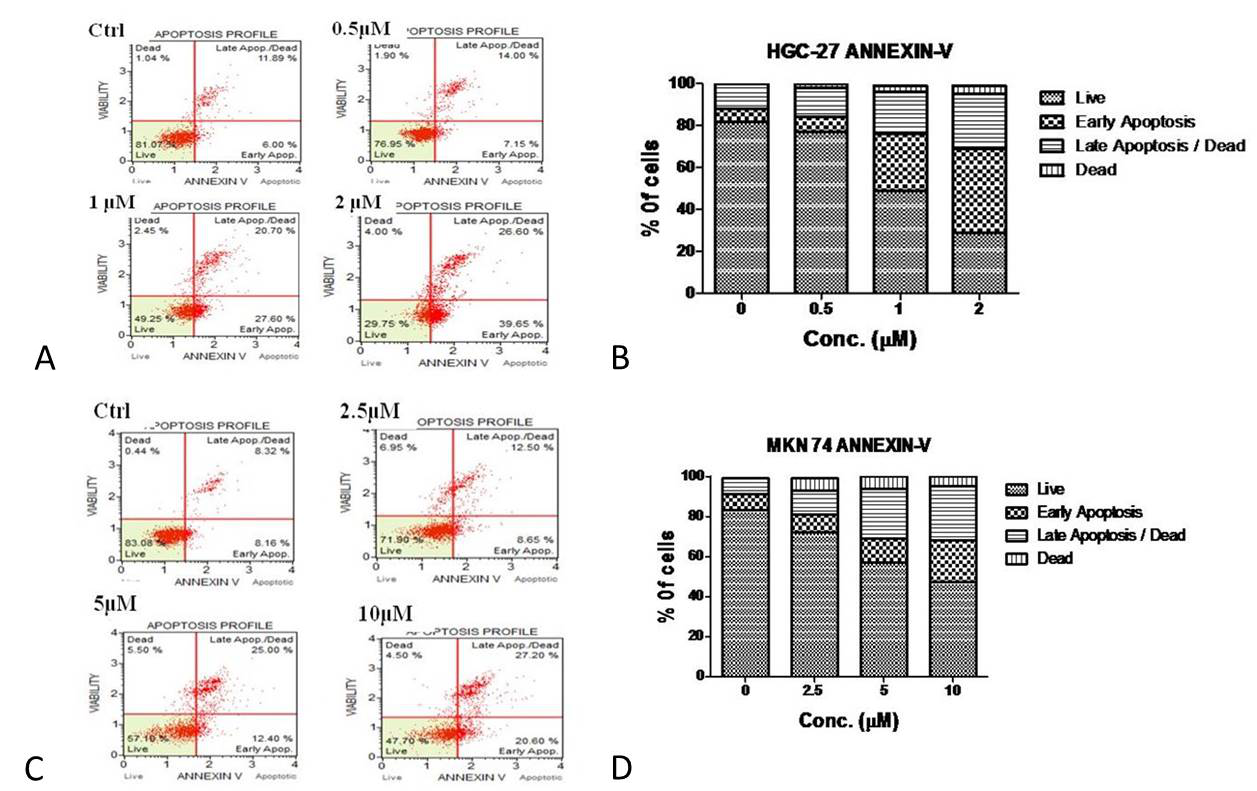 Figure 3. Apoptotic profile was determined by Annexin V assay (A) and (C) Apoptotic cell profile after exposure of indicated concentrations of niclosamide in HGC-27(0 µM, 0.5 µM, 1 µM, and 2 µM) , MKN-74 (0 µM, 2.5 µM, 5 µM, and 10 µM) respectively. (B) and (D) The graphical reperesentation of live, early, late apoptotic, dead cell populations in HGC-27, MKN-74 after treatment respectively. Data represent one of three independent experiments performed.
Figure 3. Apoptotic profile was determined by Annexin V assay (A) and (C) Apoptotic cell profile after exposure of indicated concentrations of niclosamide in HGC-27(0 µM, 0.5 µM, 1 µM, and 2 µM) , MKN-74 (0 µM, 2.5 µM, 5 µM, and 10 µM) respectively. (B) and (D) The graphical reperesentation of live, early, late apoptotic, dead cell populations in HGC-27, MKN-74 after treatment respectively. Data represent one of three independent experiments performed.
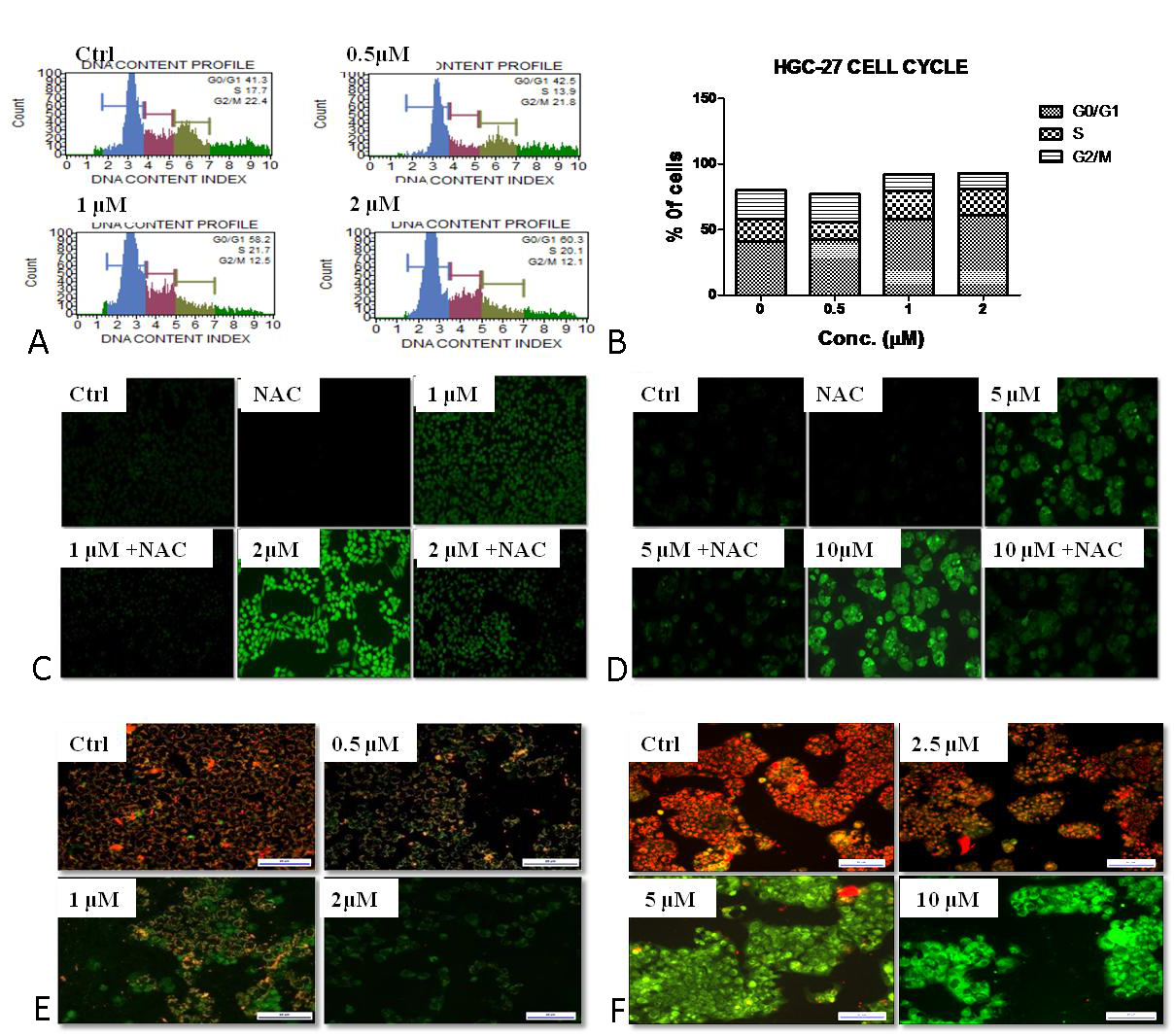 Figure 4. Effect of Niclosamide on cell cycle progression, reactive oxygen species, and mitochondrial membrane potential. (A) Cell cycle analysis in HGC-27 cells after treatment with 0 µM, 0.5 µM, 1 µM, and 2 µM concentrations of Niclosamide by Muse TM cell analyzer. (B) Cell population in each phase of cell cycle is represented in graph. Data represent one of triplicate experiments. (C), (D) Intracellular ROS determination by DCFDA method. Gastric cancer cells were treated with (1 µM and 2 µM) in HGC-27, (5 µM and10 µM) of niclosamide in MKN-74 cells for 3h and pretreated with 3 mM N- acetyl cysteine. (E), (F) Mitochondrial membrane potential assessment by JC-1. HGC-27, MKN-74 cells were exposed for (0 µM, 0.5 µM, 1 µM, and 2 µM), (0 µM, 2.5 µM, 5 µM, and 10 µM) concentrations of Niclosamide for 48 h.
Figure 4. Effect of Niclosamide on cell cycle progression, reactive oxygen species, and mitochondrial membrane potential. (A) Cell cycle analysis in HGC-27 cells after treatment with 0 µM, 0.5 µM, 1 µM, and 2 µM concentrations of Niclosamide by Muse TM cell analyzer. (B) Cell population in each phase of cell cycle is represented in graph. Data represent one of triplicate experiments. (C), (D) Intracellular ROS determination by DCFDA method. Gastric cancer cells were treated with (1 µM and 2 µM) in HGC-27, (5 µM and10 µM) of niclosamide in MKN-74 cells for 3h and pretreated with 3 mM N- acetyl cysteine. (E), (F) Mitochondrial membrane potential assessment by JC-1. HGC-27, MKN-74 cells were exposed for (0 µM, 0.5 µM, 1 µM, and 2 µM), (0 µM, 2.5 µM, 5 µM, and 10 µM) concentrations of Niclosamide for 48 h.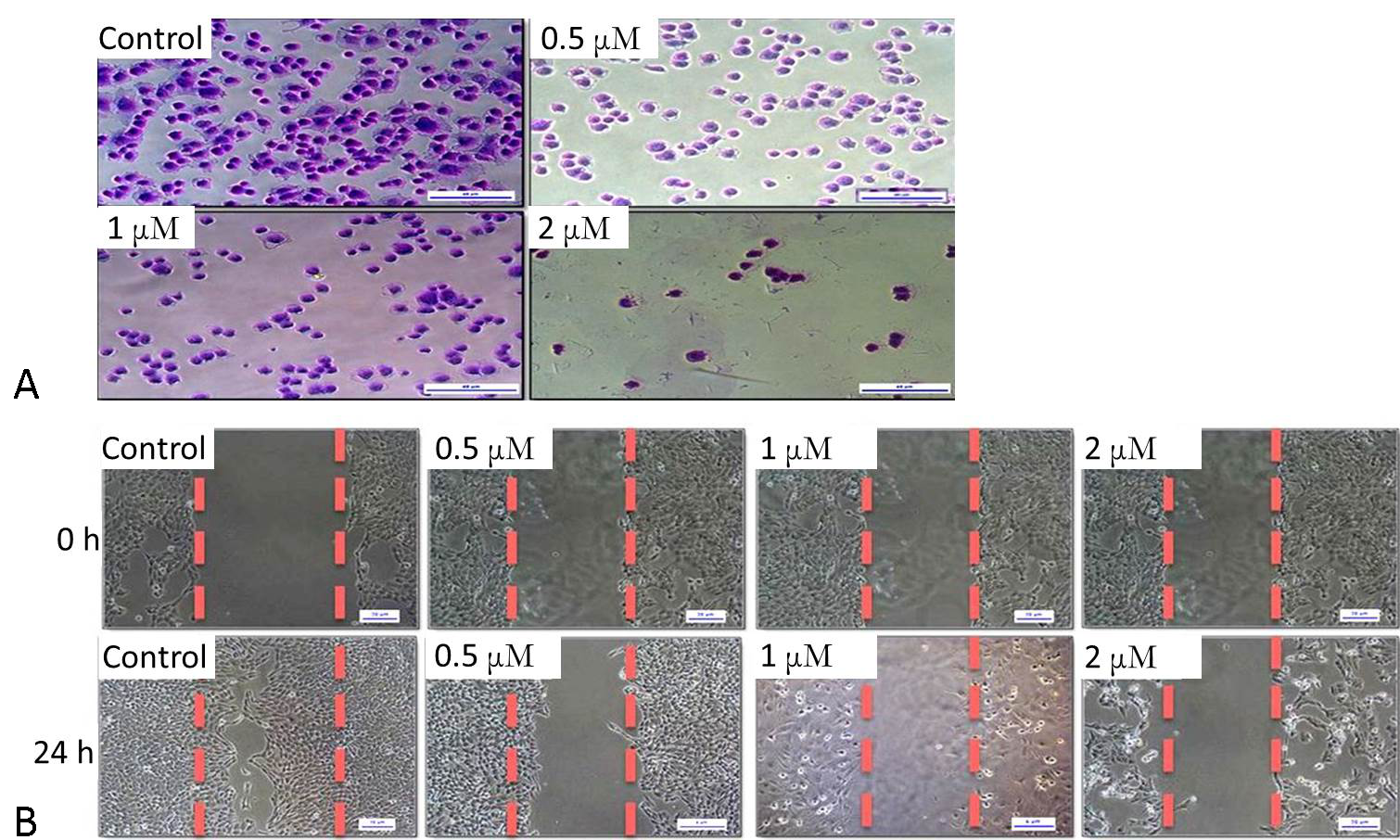 Figure 5. Niclosamide inhibits gastric cancer cell adhesion, migration. HGC-27 cells were exposed to mentioned concentrations (0 µM, 0.5 µM, 1 µM, and 2 µM) of Niclosamide. (A) Cell adhesion assay after exposure to Niclosamide for 1hr. (B) Scratch wound healing assay after 24 hrs of treatment period.
Figure 5. Niclosamide inhibits gastric cancer cell adhesion, migration. HGC-27 cells were exposed to mentioned concentrations (0 µM, 0.5 µM, 1 µM, and 2 µM) of Niclosamide. (A) Cell adhesion assay after exposure to Niclosamide for 1hr. (B) Scratch wound healing assay after 24 hrs of treatment period.
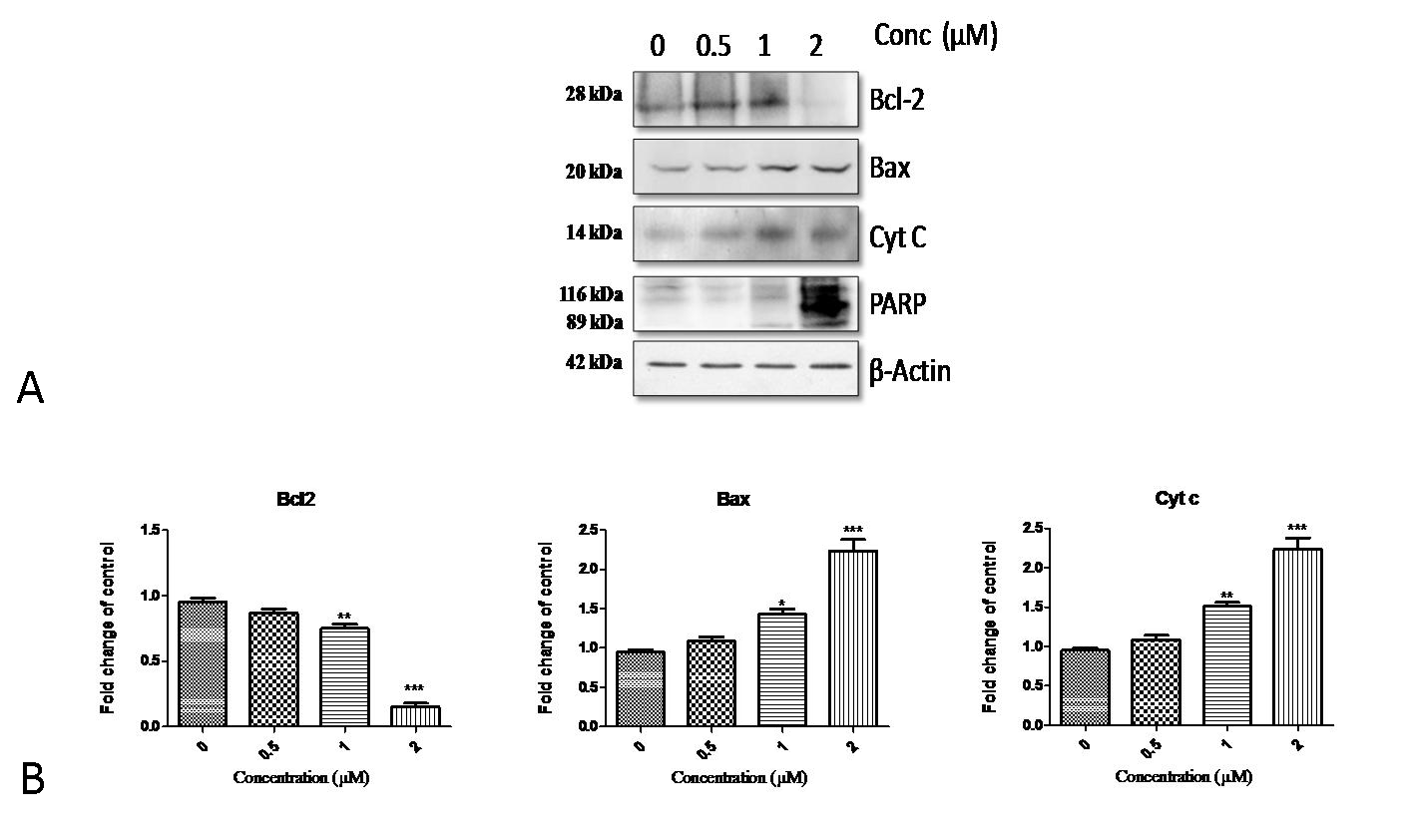 Figure 6. Effect of Niclosamide on the apoptotic protein levels. (A) Representative western blots of Bax, Bcl-2, Cyt C, PARP proteins in HGC-27. HGC-27 cells were treated with Niclosamide (0 µM, 0.5 µM, 1 µM, and 2 µM) were harvested and total cell lysates were prepared. Total 40 µg of protein was subjected to SDS-PAGE and then proceeded for western blotting analysis and chemiluminescence detection. Equal protein loading was confirmed by restriping the blots and again probing with β-actin as internal control. The western blots shown here are representative of triplicate blots with similar results. Band intensities are measured by Image J program. (B) Relative change of proteins Bcl2, Bax, Cyt C after treatment with respect to control are represented in histograms. Data are represented as Mean ± Standard Error Mean (SEM). ***P < 0.001, **P < 0.01 and *P < 0.05 vs. control.
Figure 6. Effect of Niclosamide on the apoptotic protein levels. (A) Representative western blots of Bax, Bcl-2, Cyt C, PARP proteins in HGC-27. HGC-27 cells were treated with Niclosamide (0 µM, 0.5 µM, 1 µM, and 2 µM) were harvested and total cell lysates were prepared. Total 40 µg of protein was subjected to SDS-PAGE and then proceeded for western blotting analysis and chemiluminescence detection. Equal protein loading was confirmed by restriping the blots and again probing with β-actin as internal control. The western blots shown here are representative of triplicate blots with similar results. Band intensities are measured by Image J program. (B) Relative change of proteins Bcl2, Bax, Cyt C after treatment with respect to control are represented in histograms. Data are represented as Mean ± Standard Error Mean (SEM). ***P < 0.001, **P < 0.01 and *P < 0.05 vs. control.
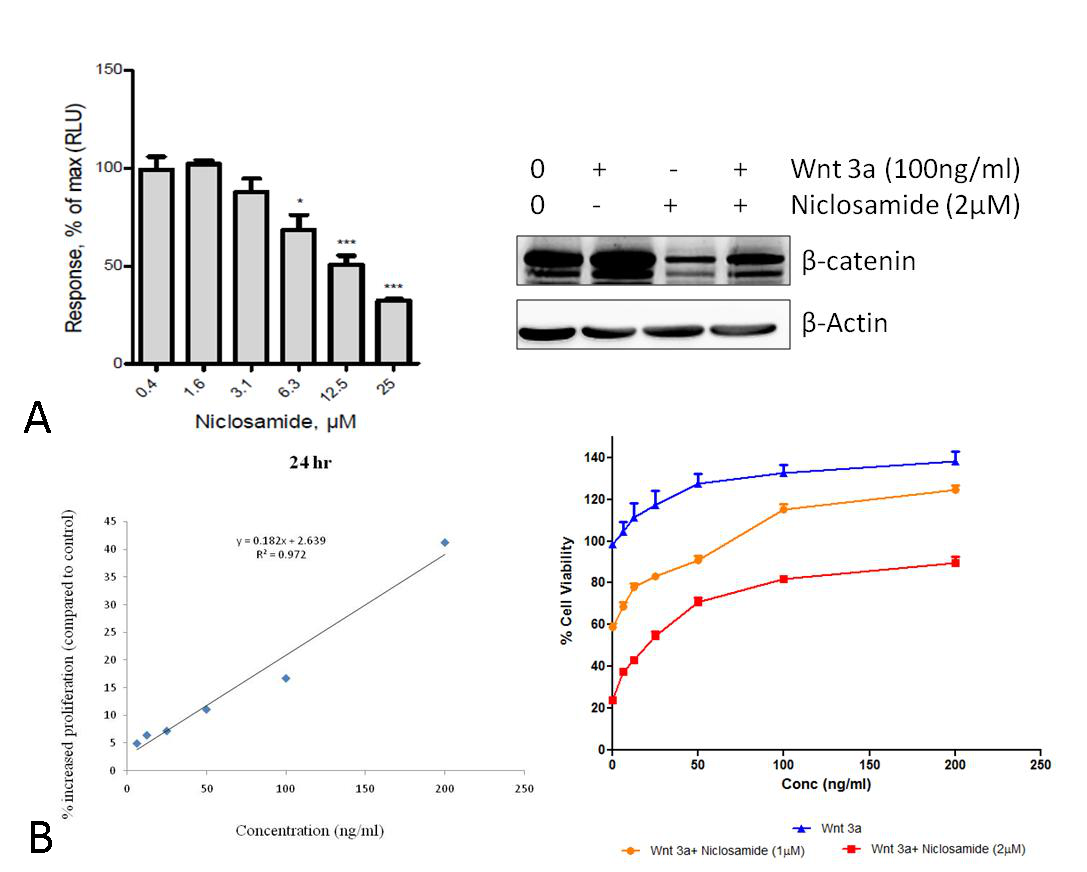 Figure 7. Effect of Niclosamide on Wnt/ β-catenin pathway. (A) TopFlash Luciferase reporter assay. (B) Effect of Niclosamide on β-catenin in wnt3a stimulated HGC-27 cells. HGC-27 cells were pretreated with wnt3a 100ng/ml and treated with Niclosamide 2 µM Cells were harvested and total cell lysates were collected and 40 µg protein was loaded in SDS-PAGE and western blotting analysis was performed. Data represent one of three independent experiments with similar results. (C) Graphical representation of effect of Wnt3a ligand on cell proliferation of HGC-27 cell line: Cells were stimulated with Wnt3a ligand (200ng/ml) stimulation for 24 h (D) Graph showing the antiproliferation effect of niclosamide in HGC-27 stimulated with wnt3a at 100, 200 ng/ml for 24h.
Figure 7. Effect of Niclosamide on Wnt/ β-catenin pathway. (A) TopFlash Luciferase reporter assay. (B) Effect of Niclosamide on β-catenin in wnt3a stimulated HGC-27 cells. HGC-27 cells were pretreated with wnt3a 100ng/ml and treated with Niclosamide 2 µM Cells were harvested and total cell lysates were collected and 40 µg protein was loaded in SDS-PAGE and western blotting analysis was performed. Data represent one of three independent experiments with similar results. (C) Graphical representation of effect of Wnt3a ligand on cell proliferation of HGC-27 cell line: Cells were stimulated with Wnt3a ligand (200ng/ml) stimulation for 24 h (D) Graph showing the antiproliferation effect of niclosamide in HGC-27 stimulated with wnt3a at 100, 200 ng/ml for 24h.
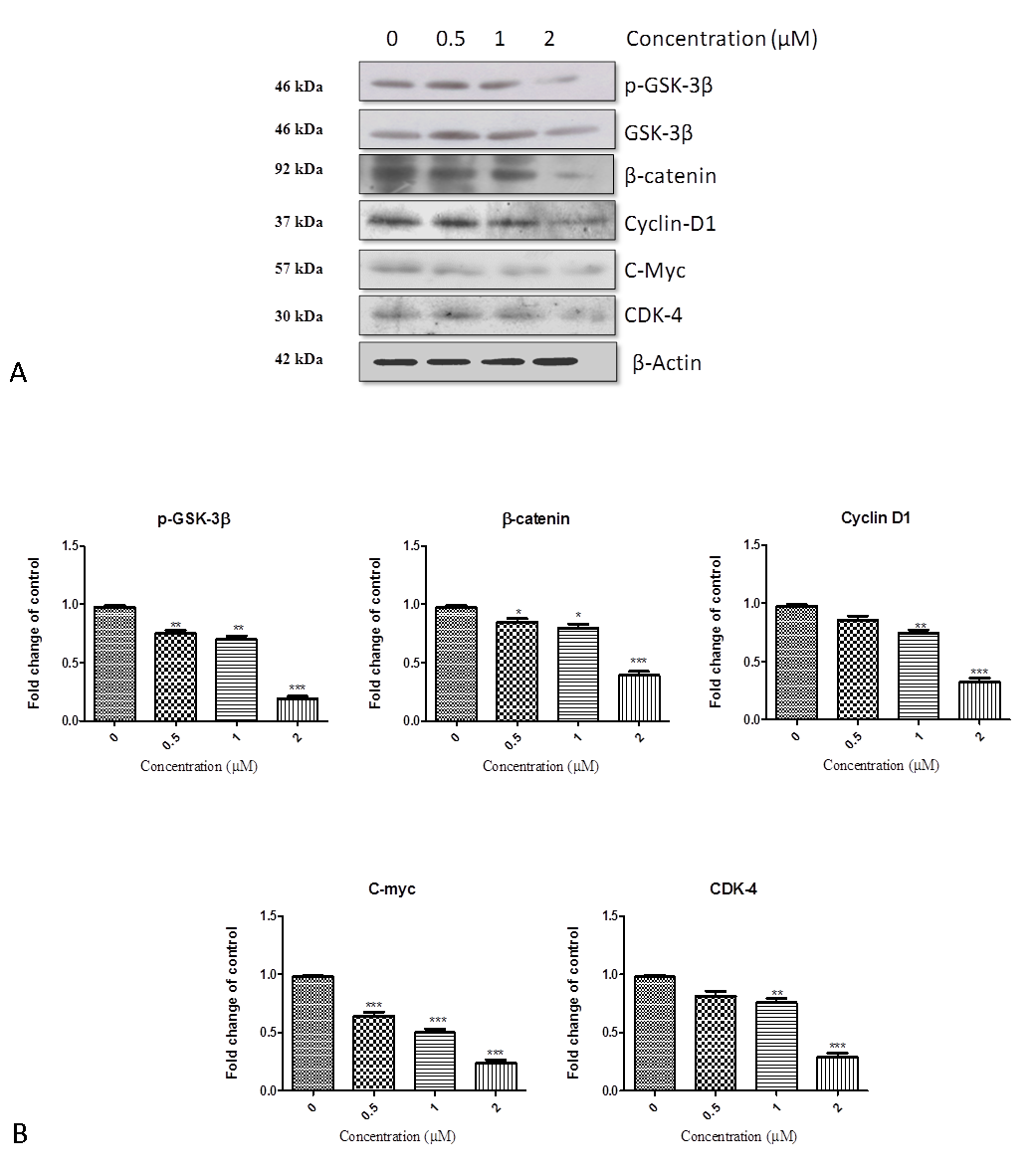 Figure 8. Effect of Niclosamide on the downstream targets of Wnt/ β-catenin pathway; (A) Representative western Blots of P-GSK-3β, GSK-3β, β- catenin, Cyclin-D1, C-Myc, CDK-4 proteins in HGC-27. HGC-27 cells were treated with niclosamide (0 µM, 0.5 µM, 1 µM, and 2 µM) were harvested and total cell lysates were prepared. Total 40 µg of protein was subjected to SDS-PAGE followed by western bloting analysis. Equal protein loading was confirmed by restriping the blots and again probing with β-actin. The western blots shown here are representative of triplicate blots with similar results. Band intensities are measured by Image J program (B) Relative change of proteins P-GSK-3β, GSK-3β, β –catenin, Cyclin-D1, C-Myc, CDK-4 after treatment with respect to control are represented in histograms. Data are Mean ± SEM. ***P < 0.001, **P < 0.01 and *P < 0.05 vs. control.
Figure 8. Effect of Niclosamide on the downstream targets of Wnt/ β-catenin pathway; (A) Representative western Blots of P-GSK-3β, GSK-3β, β- catenin, Cyclin-D1, C-Myc, CDK-4 proteins in HGC-27. HGC-27 cells were treated with niclosamide (0 µM, 0.5 µM, 1 µM, and 2 µM) were harvested and total cell lysates were prepared. Total 40 µg of protein was subjected to SDS-PAGE followed by western bloting analysis. Equal protein loading was confirmed by restriping the blots and again probing with β-actin. The western blots shown here are representative of triplicate blots with similar results. Band intensities are measured by Image J program (B) Relative change of proteins P-GSK-3β, GSK-3β, β –catenin, Cyclin-D1, C-Myc, CDK-4 after treatment with respect to control are represented in histograms. Data are Mean ± SEM. ***P < 0.001, **P < 0.01 and *P < 0.05 vs. control.
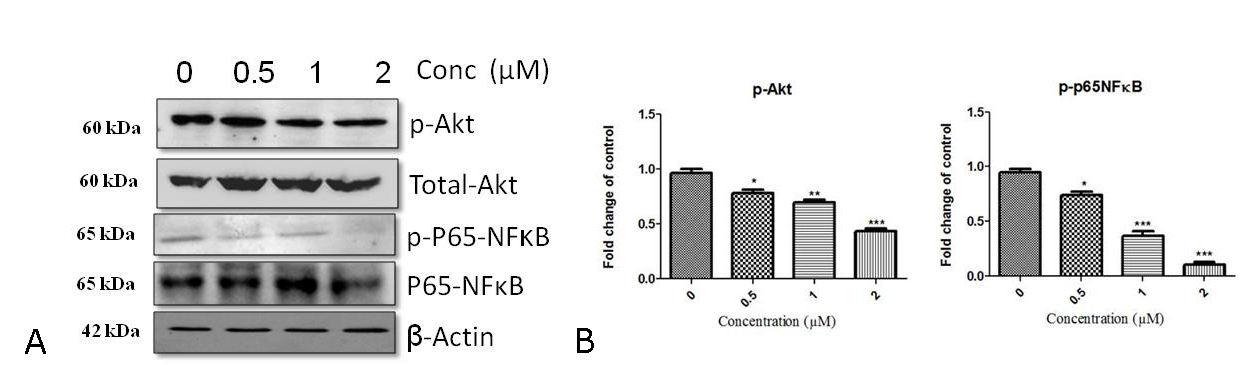 Figure 9. Effect of Niclosamide on Akt/ NF-κB axis (A) Effect of Niclosamide on p-Akt, Total- Akt, p-p65-NF-κB , P65-NF-κB levels (B) Histograms showing fold change of proteins p-Akt, p-p65 NFκB to control. HGC-27 cells were treated with indicated concentrations (0 µM, 0.5 µM, 1 µM, and 2 µM) of Niclosamide Cells were harvested and total cell cysates were collected and 40 µg protein was loaded in SDS-PAGE and western blotting analysis was performed. Data represent one of three independent experiments with similar results.
Figure 9. Effect of Niclosamide on Akt/ NF-κB axis (A) Effect of Niclosamide on p-Akt, Total- Akt, p-p65-NF-κB , P65-NF-κB levels (B) Histograms showing fold change of proteins p-Akt, p-p65 NFκB to control. HGC-27 cells were treated with indicated concentrations (0 µM, 0.5 µM, 1 µM, and 2 µM) of Niclosamide Cells were harvested and total cell cysates were collected and 40 µg protein was loaded in SDS-PAGE and western blotting analysis was performed. Data represent one of three independent experiments with similar results.
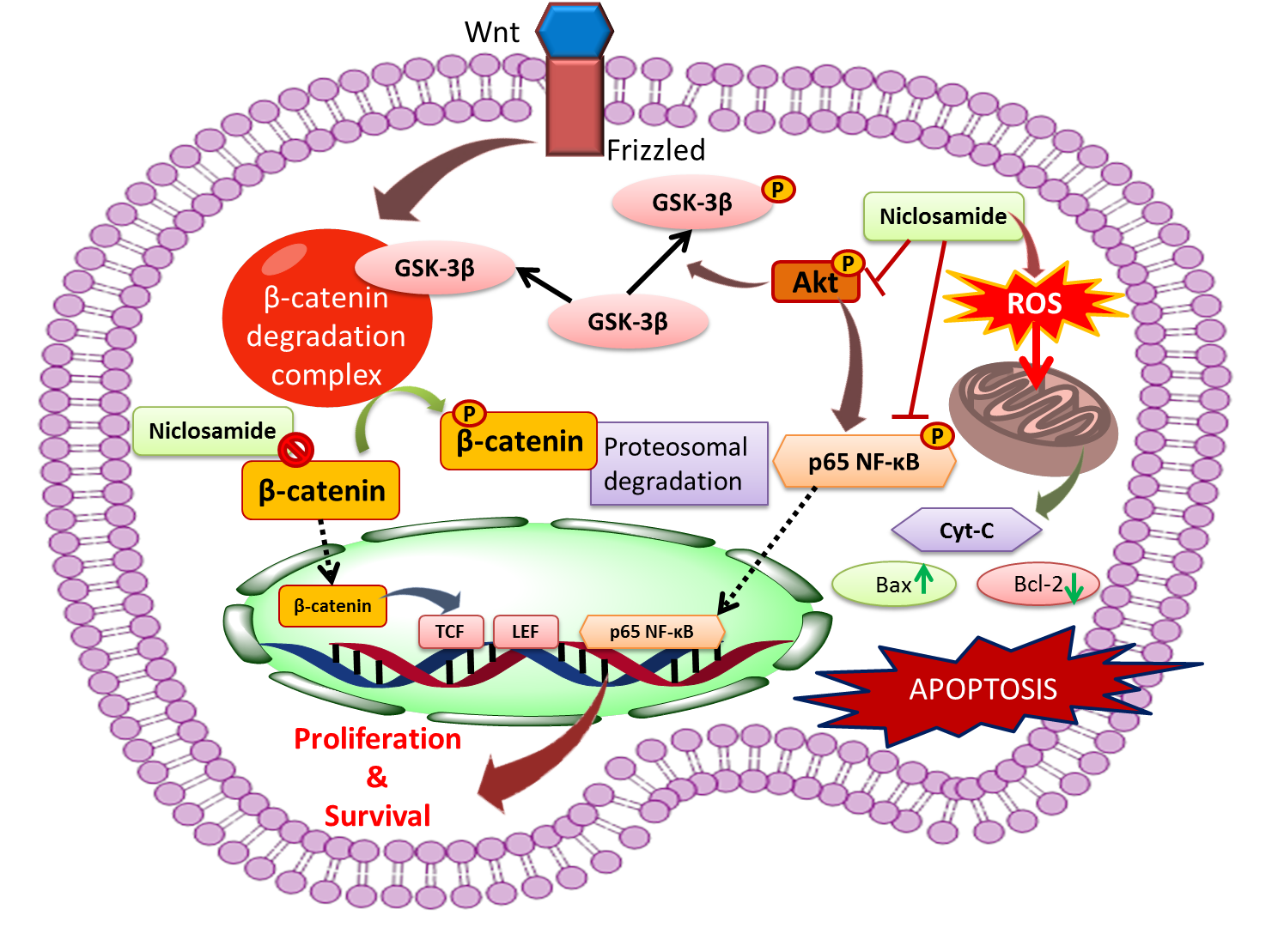 Figure 10. Proposed mechanism for antitumor activity of Niclosamide in gastric cancer cells: Niclosamide inhibited accumulation of β-catenin, a critical hall mark of activated Wnt/ β-catenin signaling and nuclear transcription of its target genes such as Cyclin-D, c-myc that are involved in cell proliferation, survival. Niclosamide possibly suppressed Akt phosphorylation and thereby inhibition of GSK-3β in the APC complex and Akt inactivation also impeded Phosphorylation of p65NF-kB, a transcription factor involved in cell survival. Furthermore, Niclosamide also potently triggered apoptosis by induction of reactive oxygen species thereby disrupting ∆ψm .Consequently this leads to release of Cytochrome c , Bax , a Bcl-2 family pro-apoptotic factor and decrease in anti apoptotic factor Bcl-2 levels that eventually results in apoptosis.
Figure 10. Proposed mechanism for antitumor activity of Niclosamide in gastric cancer cells: Niclosamide inhibited accumulation of β-catenin, a critical hall mark of activated Wnt/ β-catenin signaling and nuclear transcription of its target genes such as Cyclin-D, c-myc that are involved in cell proliferation, survival. Niclosamide possibly suppressed Akt phosphorylation and thereby inhibition of GSK-3β in the APC complex and Akt inactivation also impeded Phosphorylation of p65NF-kB, a transcription factor involved in cell survival. Furthermore, Niclosamide also potently triggered apoptosis by induction of reactive oxygen species thereby disrupting ∆ψm .Consequently this leads to release of Cytochrome c , Bax , a Bcl-2 family pro-apoptotic factor and decrease in anti apoptotic factor Bcl-2 levels that eventually results in apoptosis.
Niclosamide, a salicylanilide derivative, is an FDA approved, anthelmintic drug and has been used widely used for the treatment of tapeworm infections for almost 50 years. Besides, it is an effective and commonly used molluscicide for water treatment in the control of schistosomiasis [35]. Niclosamide uncouples oxidative phosphorylation and stimulates adenosine triphosphatase activity in mitochondria of cestodes [7]. Recently, several high throughput screening programs identified the anti-tumor activity of Niclosamide in many cancers such as breast, prostate, ovarian cancers and glioblastoma. These reports suggested that Niclosamide modulates multiple signaling pathways such as STAT3, mTORC1, and Wnt/ β-catenin signalling [8, 9, 10, 11]. Further, inhibition of cancer stem cells, a- subpopulation of tumor cells that exhibit self- renewal property and resistance to therapy, could be another important mechanism by which Niclosamide exhibits an anti-cancer effect.
In this study, we found that Niclosamide strongly inhibited the viability of gastric cancer cells in vitro (Fig. 1A). The results were comparable to the effects induced by salinomycin, a polyether ionophore antibiotic that has shown anti-tumor activity in different types of human cancers, most likely by interfering with the Wnt/β-catenin signaling pathway and other cancer stem cell pathways. However, salinomycin has reported to associate with neurotoxicity and has a narrow therapeutic index, and its clinical use has been limited [36]. In contrast, we and others have shown that Niclosamide is 5 fold more potent than salinomycin in gastric cancer and was found to be safer in normal cells [37]. Niclosamide arrested cell cycle progression at the G0/G1 phase in a dose-dependent manner. Cyclin D1 and CDK4 are essential for the G1to S phase progression in cell cycle [38]. Niclosamide markedly decreased both these proteins in a dose-dependent manner. These results are in line with a previous report that showed that Niclosamide arrested the cell cycle of the human glioblastoma cell line also in the G0/G1 Phase [39].
Apoptosis plays a pivotal role in the anti-tumor mechanism of many anti-cancer drugs. Apoptotic cells show some distinct morphological features such as membrane blubbing, chromatin condensation, nuclear fragmentation, and apoptotic body formation [40] and Niclosamide treated gastric cancer cells exhibited all these characteristics in a dose-dependent manner.
The generation of reactive oxygen species is a promising approach in cancer therapy [41]. The increased reactive oxygen species often decrease mitochondrial aerobic ATP production and disrupt mitochondrial membrane potential by oxidation of mitochondrial membrane proteins. A subsequent release of cytochrome c from the mitochondria triggers the apoptotic process [42].In our study, Niclosamide elevated reactive oxygen species generation and disrupted mitochondrial membrane potential in a concentration-dependent manner. Increased levels of cytochrome C are also clearly evident in the Niclosamide treated cells suggesting mitochondrial involvement in the anti-cancer mechanism of Niclosamide. Our findings are supported by another report showing that Niclosamide uncoupled oxidative phosphorylation and caused a loss of mitochondrial membrane potential in multiple myeloma cells[43].
Metastasis of tumor cells is the predominant cause of cancer-related death and morbidity. It is a multistep biological process that involves initial migration, invasion, intravasation, followed by extravasation and colonization in the distinct sites [44]. Here we reported that Niclosamide markedly inhibited gastric cancer cell adhesion and migration in a dose-dependent manner, implying its anti-metastatic potential in the treatment of gastric cancer.
Bcl-2 family proteins consist of both pro- and anti-apoptotic factors, primarily regulate the mitochondrial outer membrane permeabilization and thus control the commitment of cells towards intrinsic apoptotic pathway [33]. Niclosamide enhanced Bax to Bcl-2 ratio and increased PARP-1 level in treated gastric cancer cells when compared with control cells indicating the possible involvement of the intrinsic apoptotic pathway in its mechanism of anti-cancer activity.
Wnt/ β-catenin signaling pathway plays a central role in mammalian development processes such as cell proliferation, cell fate determination, cell polarity and migration, and also in tumorigenesis. Aberrational activation of Wnt signaling and mutations in various components of this pathway have been implicated in various types of tumors such as colorectal, breast, prostate, gastric, and melanoma. Enhanced β-catenin levels are typical hallmarks of the activated canonical Wnt signaling pathway, occur due to impeded phosphorylation by APC complex and inhibited proteasomal degradation. Thus accumulated β-catenin enters the nucleus and interacts with LEF/TCF forming a complex and activates downstream targets involved in cell proliferation such as cyclin D, c-Myc [5]. In our study, Niclosamide potently downregulated GSK-3β levels, β -catenin mediated transcriptional activity, total β-catenin levels, and downstream target proteins in treated cells. This is in agreement with previous literature in which Niclosamide inhibited Wnt / β-catenin signaling by promoting Frizzled receptor internalization, LRP6 degradation, thereby inhibiting β-catenin accumulation and its transcriptional activity in colorectal, breast and prostate tumors [10, 13, 45].
Akt kinase, an important component in the PI3K/AKT/mTOR pathway, also acts as an important element in the Wnt / β-catenin signaling pathway. Upon activation of Wnt signaling, Akt gets phosphorylated and activated through dishelleved protein, and thereby it phosphorylates and deactivates the GSK-3 β in the APC complex, thus stabilizes β-catenin [30]. In the present study, we examined the effect of Niclosamide on Wnt signaling mediated Akt activity and its downstream target GSK-3β, a critical component in the APC complex. Our results show that Niclosamide significantly impeded phosphorylation of Akt and its substrate GSK-3β activity in HGC-27 cells. NF-κB family transcription factors are another subset of substrates for Akt kinase that play a pivotal role in inflammation, cell survival and tumorigenesis, and evasion of apoptosis [32, 34]. In this study, Niclosamide completely suppressed phosphorylated P65 NF-κB form, one of the critical components of the NF-κB pathway.
In conclusion, our study demonstrated that Niclosamide has potent anti-tumor activity in gastric cancer cells through inhibition of Wnt/ β-catenin and also its associated Akt, NF-kB signaling pathways as well as generation of reactive oxygen species. Since this pathway is highly involved in the cancer stem cells, further work should be done on the anti-cancer effect of Niclosamide on gastric cancer stem cells. Hence this report emphasizes that Niclosamide has the potential to become a promising anti-cancer agent in the treatment of human gastric cancer and further warrants the initiation of clinical trials in gastric cancer patients.
Acknowledgments
The authors are thankful to the Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Government of India, for providing funds. VGMN thanks DST-SERB for the Young Scientists grant. The authors would like to express thanks to the anonymous reviewers for their critical suggestions to improve the language and clarity of the manuscript.
Funding
This research did not receive any specific financial support from public or commercial agencies.
Author contributions
MKJ, SK, and SS carried out in-vitro studies and performed statistical analysis. SNP drafted manuscript and helped in statistical analysis. VGMN, SU, and VLK conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
All authors declare no competing interests.
Ethical statement
There is no ethical statement as we haven't included any animals or human subjects in this study.
- Nagini S: Carcinoma of the stomach: A review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J Gastrointest Oncol 2012, 4(7): 156-169.
- Cervantes A, Roda D, Tarazona N, Roselló S, Pérez-Fidalgo J: Current questions for the treatment of advanced gastric cancer. Cancer Treat Rev 2013, 39(1): 60-67.
- Meyer H-J, Wilke H: Treatment strategies in gastric cancer. Dtsch Arztebl Int 2011, 108(41): 698-706.
- Nusse R, Clevers H: Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169(6): 985-999.
- Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W, Park JK, Wang H, Peng Y, Jiang W: Wnt/β-catenin signaling pathway as novel cancer drug targets. Curr Cancer Drug Targets 2004, 4(8): 653-671.
- Jin G, Wong ST: Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today 2014, 19(5): 637-644.
- Sekhon BS: Repositioning drugs and biologics: Retargeting old/existing drugs for potential new therapeutic applications. J Pharm Educ Res 2013, 4(4): 1-15.
- Fonseca BD, Diering GH, Bidinosti MA, Dalal K, Alain T, Balgi AD, Forestieri R, Nodwell M, Rajadurai CV, Gunaratnam C: Structure-activity analysis of Niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem 2012, 287(21): 17530-17545.
- Ren X, Duan L, He Q, Zhang Z, Zhou Y, Wu D, Pan J, Pei D, Ding K: Identification of Niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. ACS Med Chem Lett 2010, 1(9): 454-459.
- Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y: Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. Plos One 2011, 6(12): e29290.
- Jin YL, Lin Z, Ding K, Li J, Du X, Chen C, Sun XY, Wu YB, Zhou J, and Pan JX: Antineoplastic Mechanisms of Niclosamide in Acute Myelogenous Leukemia Stem Cells: Inactivation of the NF-κB Pathway and Generation of Reactive Oxygen Species. Cancer Res 2010, 70(6): 2516-2527.
- Wang YC, Chao TK, Chang CC, Yo YT, Yu MH, Lai HC: Drug screening identifies Niclosamide as an inhibitor of breast cancer stem-like cells. PLoS One 2013, 8(9): e74538.
- Naidu V, Bandari UM, Giddam AK, Babu KRD, Ding J, Babu KS, Ramesh B, Pragada RR, Gopalakrishnakone P: Apoptogenic activity of ethyl acetate extract of leaves of Memecylon edule on human gastric carcinoma cells via mitochondrial dependent pathway. Asian Pac J Trop Med 2013, 6(5): 337-45.
- Ribble D, Goldstein NB, Norris DA, Shellman YG: A simple technique for quantifying apoptosis in 96-well plates. BMC biotechnology 2005, 5: 12.
- Kloesch B, Becker T, Dietersdorfer E, Kiener H, Steiner G: Anti-inflammatory and apoptotic effects of the polyphenol curcumin on human fibroblast-like synoviocytes. Int Immunopharmacol 2013, 15(2): 400-405.
- Mallavadhani UV, Prasad CV, Shrivastava S, Naidu V: Synthesis and anti-cancer activity of some novel 5, 6-fused hybrids of juglone based 1, 4-naphthoquinones. Eur J Med Chem 2014, 83: 84-91.
- Kim HJ, Suh HJ, Kim JH, Park S, Joo YC, Kim JS: Antioxidant activity of glyceollins derived from soybean elicited with Aspergillus sojae. J Agric Food Chem 2010, 58(22): 11633-11638.
- Ovadje P, Chatterjee S, Griffin C, Tran C, Hamm C, Pandey S: Selective induction of apoptosis through activation of caspase-8 in human leukemia cells (Jurkat) by dandelion root extract. J Ethnopharmacol 2011, 133(1): 86-91.
- Chen Y: Scratch Wound Healing Assay. Bio-protocol 2012, 2(5): e100.
- Humphries MJ: Cell adhesion assays. Molecular biotechnology 2001, 18(1): 57-61.
- Shrivastava S, Kulkarni P, Thummuri D, Jeengar MK, Naidu VGM, Alvala M, Redddy GB, Ramakrishna S: Piperlongumine, an alkaloid causes inhibition of PI3 K/Akt/mTOR signaling axis to induce caspase-dependent apoptosis in human triple-negative breast cancer cells. Apoptosis 2014, 19(7): 1148-1164.
- Koval A, Vlasov P, Shichkova P, Khunderyakova S, Markov Y, Panchenko J, Volodina A, Kondrashov F, Katanaev V: Anti-leprosy drug clofazimine inhibits growth of triple-negative breast cancer cells via inhibition of canonical Wnt signaling. Biochem Pharmacol 2014, 87(4): 571-578.
- Mosimann C, Hausmann G, Basler K: The role of Parafibromin/Hyrax as a nuclear Gli/Ci-interacting protein in Hedgehog target gene control. Mech Dev 2009, 126(5-6): 394-405.
- Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R: A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal Biochem 2000, 282(1): 158-161.
- Indran IR, Tufo G, Pervaiz S, Brenner C: Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta 2011, 1807(6): 735-745.
- Ly JD, Grubb D, Lawen A: The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis 2003, 8(2): 115-128.
- Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP: Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Lett 2000, 475(3): 267-272.
- Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F: Mitochondria-ros cross-talk in the control of cell death and aging. J Signal Transduct 2012, 2012(329635): 1-17.
- Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C, Groden J, Lowy AM: β-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res 2002, 62(12): 3503-3506.
- Fukumoto S, Hsieh CM, Maemura K, Layne MD, Yet SF, Lee KH, Matsui T, Rosenzweig A, Taylor WG, Rubin JS: Akt participation in the Wnt signaling pathway through Dishevelled. J Biol Chem 2001, 276(20): 17479-17483.
- Vara JÁF, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M: PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 2004, 30(2): 193-204.
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JPY, Baldwin AS: Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev 2008, 22(11): 1490-1500.
- Danial NN: BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res 2007, 13(24): 7254-7263.
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB: NF-κB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature 1999, 401(6748): 82-85.
- Pan KF, Liu WG, Zhang L, You WC, Lu YY: Mutations in components of the Wnt signaling pathway in gastric cancer. World J Gastroenterol 2008, 14(10): 1570-1574.
- Boehmerle W, Muenzfeld H, Springer A, Huehnchen P, Endres M: Specific targeting of neurotoxic side effects and pharmacological profile of the novel cancer stem cell drug salinomycin in mice. J Mol Med 2014, 92(8): 889-900.
- Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y: Niclosamide Suppresses Cancer Cell Growth by Inducing Wnt Co-Receptor LRP6 Degradation and Inhibiting the Wnt/β-Catenin Pathway. PLoS One 2011, 6(12): e29290.
- Hahm ER, Singh SV: Honokiol causes G0-G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol Cancer Ther 2007, 6(10): 2686-2695.
- Wieland A, Trageser D, Gogolok S, Reinartz R, Höfer H, Keller M, Leinhaas A, Schelle R, Normann S, Klaas L: Anti-cancer effects of Niclosamide in human glioblastoma. Clin Cancer Res 2013, 9(15): 4124-4136.
- Green DR: Means to an end: apoptosis and other cell death mechanisms. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y 2012.
- Nogueira V, Hay N: Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res 2013, 19(16): 4309-4314.
- Ly JD, Grubb D, Lawen A: The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis 2003, 8(2): 115-128.
- Khanim F, Merrick B, Giles H, Jankute M, Jackson J, Giles L, Birtwistle J, Bunce C, Drayson M: Redeployment-based drug screening identifies the anti-helminthic Niclosamide as anti-myeloma therapy that also reduces free light chain production. Blood Cancer J 2011, 1(10): e39.
- Valastyan S, Weinberg RA: Tumor metastasis: molecular insights and evolving paradigms. Cell 2011, 147(2): 275-292.
- Chen M, Wang J, Lu J, Bond MC, Ren XR, Lyerly HK, Barak LS, Chen W: The anti-helminthic Niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48(43): 10267-10274.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.