
 Submit Manuscript
Submit Manuscript
Review | Open Access
QSAR-driven optimization of small-molecule inhibitors against BRAF-mutant melanoma
Xiaocui Yu1, Yi Ding2, Guiying Zhang1, Zhenyu Cao1, Yingying Zhao1, Yuwei Zhu1, Chunpu Li1, Zhenhua Fan1, Zihui Ma1, Junwei Xu1, Zexin Wang1, Wenkai Han1, Xiang Yu1, Jin Gao1, Dan Hu1
1General Station for Drug & Instrument Supervision and Control Joint Logistics Support Force PLA, Beijing 100071, China.
2Department of Pharmacy, Xijing Hospital, Fourth Military Medical University, Xi'an City 710000, Shaanxi Province, China.
Correspondence: Jin Gao and Dan Hu (General Station for Drug & Instrument Supervision and Control Joint Logistics Support Force PLA, No. 17, Fengtai West Road, Fengtai District, Beijing 100071, China; Jin Gao's Email: angelgaojin@163.com, Dan Hu's Email: hudan1006@sina.com).
Asia-Pacific Journal of Oncology 2025, 6: 70-81. https://doi.org/10.32948/ajo.2025.10.24
Received: 08 Sep 2025 | Accepted: 22 Oct 2025 | Published online: 18 Nov 2025 
Key words melanoma, quantitative structure activity relationship, BRAF mutantinhibitors, BRAF inhibitors
Melanoma arises from the un-controlled proliferation of melanocytes in the epidermal layer and represents the most severe type of cutaneous malignancy. During the course of melanoma development, multiple molecular alterations including the excessive activation of the mitogen-activated protein kinase (MAPK) growth regulatory pathway may develop and become the subject of therapeutic target [4]. Two common oncogenes that are related to melanoma are BRAF and NRAS [5]. The BRAF gene encodes the BRAF protein, a crucial component of the MAPK- pathway. This pathway comprises a sequence of intracellular proteins governing cellular expansion, programmed cell death, and differentiation. [6]. The SBRAF mutations are found in about 50% of patients with metastatic melanoma. The V600E and V600K mutations are the most prevalent types of BRAF mutations in patients with BRAF-mutant melanoma, and the former is present in 70-90 of individuals with this disease. The majority of BRAF mutations target the kinase domain of the BRAF-protein resulting in permanent activation and heightened MAPK-signaling, which all stimulate malignant development [7].
Tumors posses the ability to develop out of various genetically distinct groups of cells leading to tumor heterogeneity that characterizes cellular-heterogeneity of the tumor microenvironment (TME). One of the primary hurdles in advancing novel personalized medicine development to cure cancer is tumor heterogeneity. The inter-tumor heterogeneity of BRAF has been associated with melanoma in about 4- 25% of patients [8]. In a case report on a 49-year-old Japanese woman with metastatic BRAF-mutant melanoma, it was reported that Sanger sequencing revealed that BRAF V600E and BRAF wild are the status of the primary tumor and skin metastatic lesions, respectively, which indicates that the melanoma of the BRAF genotype.
The most common form of B-RAF mutation is V600E (valine to glutamic acid) that causes constitutive B-RAF kinase activation leading to unregulated cell replication [9]. Other less frequent mutations are V600K, V600R, V600E2, and V600D. B-RAF mutations in the V600E loci contribute more than 90% of all B-RAF mutations and V600K mutations are seen in 5 to 30 % cases. The changes result in continuous MEK/ERK signals that are favorable to tumor growth and drug resistance.
The initial purpose of B-RAF as a cancer therapeutic target was founded on a hypothesis that its activity would suppress RAS/RAF/MEK/ERK-signaling pathways and, therefore, inhibit tumor progression. Nonetheless, B-RAF inhibitors have developed resistance that hinders their sustainability. B-RAF inhibition is evaded by several resistance mechanisms that include secondary mutations, activating alternative signaling pathways, and adjustments within the tumor microenvironment.[10, 11]. With the current outburst of BRAF inhibitors, current studies are exploring combination therapy as a viable solution to the problem. A potential approach is the combination of inhibiting both B-RAF and MEK, a strategy that has demonstrated superior clinical results because of increased activation of MAPK [12]. The invention of B-RAF V600 kinase inhibitors has contributed greatly to the targeted melanoma treatment, especially against cancers initiated by B-RAF V600 mutations. These inhibitors interfere with the RAS/RAF/ MEK/ERK pathway that contributes significantly to the development and progression of tumors.
Three BRAFi/MEKi combinations are currently recommended as therapies of BRAF-mutated melanoma, cobimetinib +vemurafenib, trametinib + dabrafenib, and encorafenib + binimetinib. These three combinations are similar and active, even though there have not been direct comparative studies. Encorafenib however has clarified a longer and stronger pharmacodynamic activity over vemurafenib and dabrafenib and may have better clinical consequences [13, 14]. Targeted therapies such as vemurafenib and dabrafenib are proven to change the treatment of melanoma, have some limitations and may induce side effects (such as skin rash, fatigue, cardiovascular problems)[15]. Phytochemicals and especially natural sources are potentially useful to multi-target approaches [16]. Conversely, FDA-approved drugs like vemurafenib and dabrafenib focus on precise molecular abnormalities, which could restrict their approach to different groups of cancerous cells [17]. The conventional high throughput screening on inhibitors against BRAF-mutant melanoma is limited and it has been further transformed by an exploited approach in quantum chemistry for drug research and discovery, which is the Quantitative Structure Activity Relationship (QSAR). QSAR utilizes structural analysis to gauge chemical bioactivity and forecast the potency of novel treatment options [18].
Chemprop represents a successful application of this model; it is a graph neural network-based QSAR framework that learns directly from molecular graph, which scales a wide range of chemical space with high predictive quality and scalability. It has proved to be better or equal to conventional descriptor-based QSAR models in diverse benchmark investigations since it is able to incorporate intricate, non-linear structure impact relationships without handcrafted feature engineering. [19].
This mini review will offer a summary of the molecular pathophysiology underlying BRAF-mutant melanoma, and existing therapeutic options of the MAPK pathway, especially the use of quantitative structure activity relationship (QSAR) methodologies in the design and optimization of small molecule BRAF inhibitors. We emphasize the latest QSAR-related research, show how it is combined with other computational tools, and present the latest problems and prospects of providing better therapy of BRAF-driven melanoma. Schematic diagram of BRAF V600E-resultant signaling initiates melanoma formation and progression as shown in Figure 1.
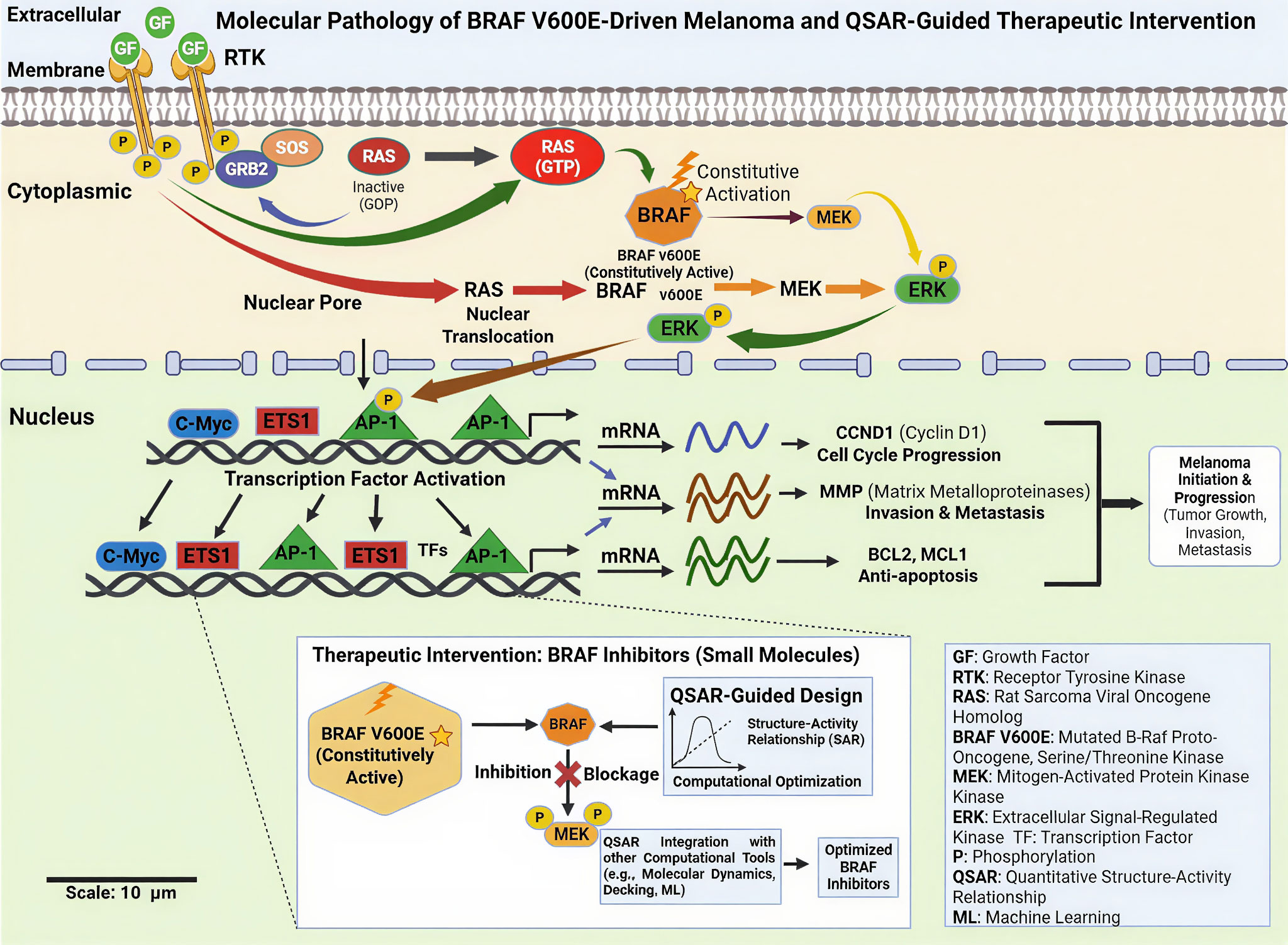 Figure 1. Schematic flow of the biochemical pathology of BRAF V600E-mediated melanoma and the therapeutic intervention guided by QSAR. GRB2/SOS-mediated RAS activation is stimulated by the growth factor binding of receptor tyrosine kinases (RTKs). Active BRAF V600E is constitutively active and does not depend on an upstream regulator, thus activating MEKERK signaling cascade in a continuous manner. ERK activation is translocated to the nucleus, which activates transcription factors (AP-1, ETS1, c-Myc) and expression of downstream genes connected to cell-cycle progression (CCND1), invasion and metastasis (MMPs), and resistance to apoptosis (BCL2, MCL1). The inset points out the QSAR-based design of small molecule BRAF inhibitors that are selective in inhibiting aberrant signaling that inhibits melanoma initiation, progression, and metastasis.
Figure 1. Schematic flow of the biochemical pathology of BRAF V600E-mediated melanoma and the therapeutic intervention guided by QSAR. GRB2/SOS-mediated RAS activation is stimulated by the growth factor binding of receptor tyrosine kinases (RTKs). Active BRAF V600E is constitutively active and does not depend on an upstream regulator, thus activating MEKERK signaling cascade in a continuous manner. ERK activation is translocated to the nucleus, which activates transcription factors (AP-1, ETS1, c-Myc) and expression of downstream genes connected to cell-cycle progression (CCND1), invasion and metastasis (MMPs), and resistance to apoptosis (BCL2, MCL1). The inset points out the QSAR-based design of small molecule BRAF inhibitors that are selective in inhibiting aberrant signaling that inhibits melanoma initiation, progression, and metastasis.
The dysregulation of various signaling pathways that interact with each other are the drivers of BRAF-mutant melanoma, with the central oncogenic axis being the mitogen-activated protein kinase/ extracellular signal-regulated kinase (MAPK/ERK) pathway [21]. The stimulation of BRAF mutations and V600E results in constitutive MAPK transmission with continuous MEK and ERK stimulation, which facilitates uncontrolled proliferation, survival, and invasion. Besides MAPK, phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR pathway is also often concomitantly activated in BRAF-mutant melanoma, whether by loss of PTEN, RTK upregulation or by adaptive resistance, leading to the heightened cell survival, metabolic reprogramming and resistance to therapy [22, 23]. Crosstalk of MAPK and PI3K/AKT signaling during melanoma cell survival allows the cells to overcome the inhibitory effect of BRAF and maintain oncogenic signaling. WNT/cadherin WNT/cadherin pathway is also contextually dependent in melanoma progression, and affects cell differentiation, immune evasion, and metastatic behavior. Also, the Hippo-YAP/TAZ dysregulation has been associated with the stimulation of melanoma cells plasticity, survival, and resistance to targeted therapy. Changes in the apoptotic signaling pathways, such as inhibition of pro-apoptotic factors and expression of the anti-apoptotic proteins also aid in the persistence of tumors. Collectively, these signaling pathways represent a highly adaptive signaling network underlying melanoma progression, therapeutic resistance and heterogeneity of the melanoma in BRAF-mutant melanoma disease [20]. The pathways involved in melanoma progression have been illustrated in Figure 2.
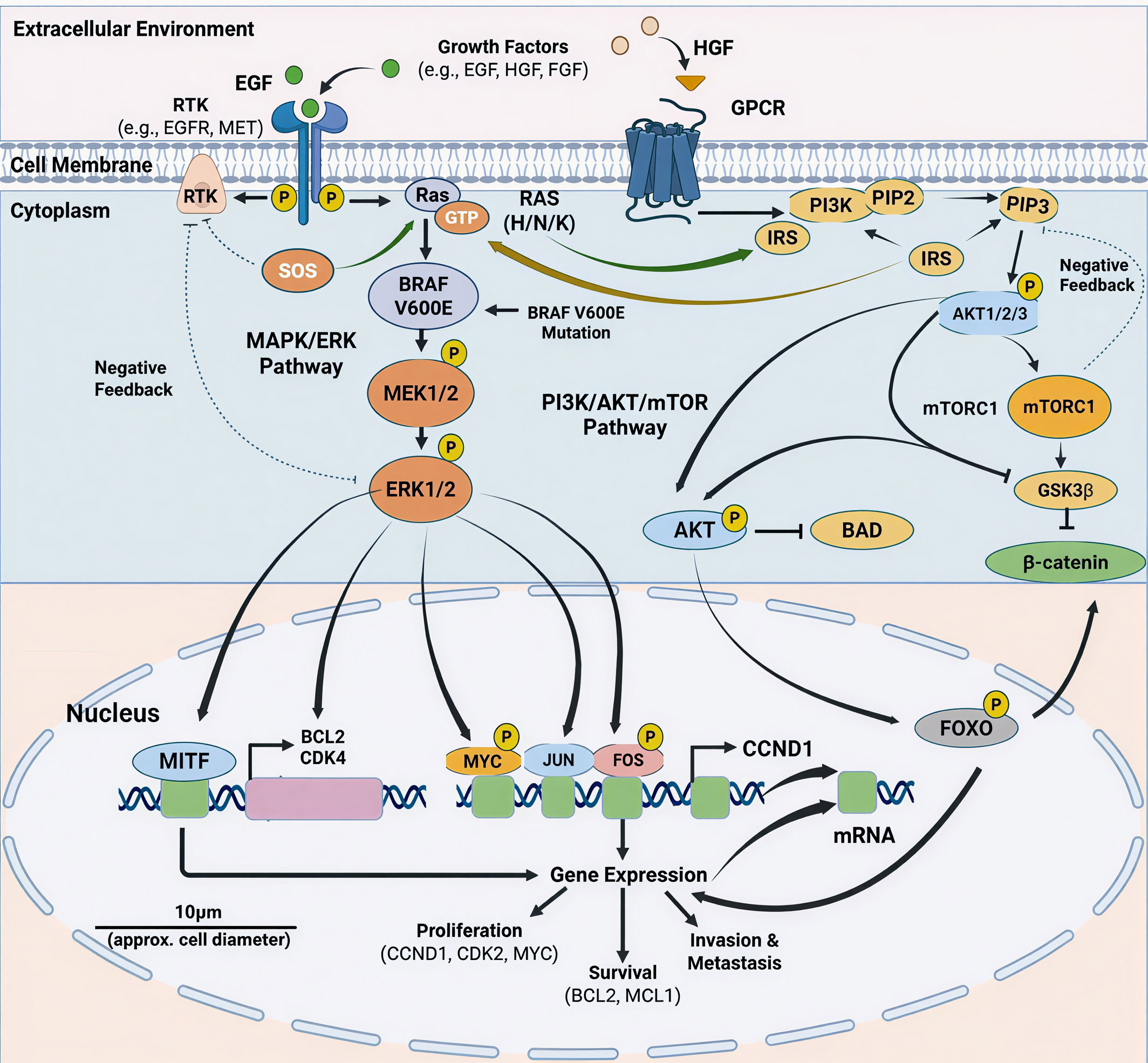 Figure 2. Combinational schematic of oncogenic signaling networks driven by the BRAF V600E mutation in melanoma with cross-talk between the MAPK/ERK and PI3K/AKT/mTOR pathways. Receptor tyrosine kinases (RTKs) or GPCRs are stimulated by growth factors (EGF, HGF, FGF) and cause the activation of RAS (H/N/K). BRAF V600E constitutively active BRAF V600E maintains phosphorylation of MEK1/2 and ERK1/2 facilitating translocation and activation of nuclear transcription factors, such as MITF, MYC, JUN and FOS. Simultaneously, the activation of PI3K leads to the creation of PIP3, which is the activator of AKT and mTORC1 signaling, controlling survival and metabolism through BAD, FOXO, GSK3 2, and 2-catenin. These joint programs promote proliferation (CCND1, CDK2), survival (BCL2, MCL1), and invasion/metastasis programs and pathways are regulated by negative feedback loops.
Figure 2. Combinational schematic of oncogenic signaling networks driven by the BRAF V600E mutation in melanoma with cross-talk between the MAPK/ERK and PI3K/AKT/mTOR pathways. Receptor tyrosine kinases (RTKs) or GPCRs are stimulated by growth factors (EGF, HGF, FGF) and cause the activation of RAS (H/N/K). BRAF V600E constitutively active BRAF V600E maintains phosphorylation of MEK1/2 and ERK1/2 facilitating translocation and activation of nuclear transcription factors, such as MITF, MYC, JUN and FOS. Simultaneously, the activation of PI3K leads to the creation of PIP3, which is the activator of AKT and mTORC1 signaling, controlling survival and metabolism through BAD, FOXO, GSK3 2, and 2-catenin. These joint programs promote proliferation (CCND1, CDK2), survival (BCL2, MCL1), and invasion/metastasis programs and pathways are regulated by negative feedback loops.
There are currently three approved combinations of BRAFi/MEKi, dabrafenib with trametinib (COMBI-d and COMBI-v studies), vemurafenib with cobimetinib (CoBRIM study), and encorafenib with binimetinib (COLUMBUS study) which showed better response rates and survival than BRAFi alone, with the encorafenib-binimetinib combination having a longer median OS [20] (Table 1). Figure 3 shows the significant signaling pathways of BRAF-mutant melanoma and how they can be targeted therapeutically. Activating BRAF mutants stimulates constitutive MAPK/ERK depolarization, which facilitates melanoma growth and survival. This pathway is blocked with selective BRAF inhibitors and MEK inhibitors, and a combination of BRAF and MEK inhibition increases therapeutic responses and decreases resistance. Interaction with the PI3K/AKT/mTOR signaling also adds to tumor survival and resistance to treatment.
|
Table 1. Pivotal targeted therapy trials influencing clinical practice in melanoma patients. |
|||||||||
|
Therapy Category |
Trial (Clinical ID) |
Phase |
Patient Population |
Treatment Arms |
Primary Endpoint |
n |
ORR (%) |
Median PFS (mo) |
Median OS (mo) |
|
Anti–CTLA-4 |
MDX010-020 (NCT00094653) |
III |
Untreated metastatic melanoma |
Ipilimumab + gp100 vs Ipilimumab vs gp100 |
OS |
676 |
6 vs 11 vs 2 |
2.8 vs 2.9 vs 2.8 |
10.0 vs 10.1 vs 6.4 |
|
Anti–PD-1 |
CM-066 (NCT01721772) |
III |
Untreated BRAF wild-type melanoma |
Nivolumab vs Dacarbazine |
OS |
418 |
40 vs 14 |
5.1 vs 2.2 |
37.5 vs 11.2 |
|
Anti–PD-1 ± CTLA-4 |
CM-067 (NCT01844505) |
III |
Untreated metastatic melanoma |
Nivo+Ipi vs Nivo vs Ipi |
PFS & OS |
945 |
58 vs 44 vs 19 |
11.5 vs 6.9 vs 2.9 |
NR |
|
Anti–PD-1 + CTLA-4 |
CM-511 (NCT02714218) |
III |
Untreated metastatic melanoma |
Nivo1+Ipi3 vs Nivo3+Ipi1 |
TRAE (Grade 3–5) |
360 |
48 vs 34 |
8.9 vs 9.9 |
NR |
|
Anti–PD-1 |
KN-006 (NCT01866319) |
III |
Melanoma ≤1 prior therapy line |
Pembrolizumab q2w vs q3w vs Ipilimumab |
PFS & OS |
834 |
34 vs 33 vs 12 |
8.4 vs 3.4 |
32.7 vs 15.9 |
|
BRAF inhibitor |
BRIM-3 (NCT01006980) |
III |
Untreated metastatic melanoma |
Vemurafenib vs DTIC |
PFS & OS |
675 |
48 vs 5 |
5.3 vs 1.6 |
13.6 vs 9.7 |
|
BRAF inhibitor |
BREAK-3 (NCT01227889) |
III |
Untreated BRAFV600E melanoma |
Dabrafenib vs DTIC |
ORR |
250 |
50 vs 6 |
6.9 vs 2.7 |
20 vs 15.6 |
|
BRAF + MEK inhibitors |
COMBI-v (NCT01597908) |
III |
Untreated BRAFV600E/K melanoma |
Dabrafenib+Trametinib vs Vemurafenib |
OS |
704 |
64 vs 51 |
11.4 vs 7.3 |
NR vs 17.2 |
|
BRAF + MEK inhibitors |
COMBI-d (NCT01584648) |
III |
Untreated BRAFV600E/K melanoma |
Dabrafenib+Trametinib vs Dabrafenib |
PFS |
423 |
69 vs 53 |
11.0 vs 8.8 |
25.1 vs 18.7 |
|
BRAF + MEK inhibitors |
CoBRIM (NCT01689519) |
III |
Untreated BRAFV600 melanoma |
Cobimetinib+Vem vs Vem+Placebo |
PFS |
495 |
68 vs 45 |
12.3 vs 7.2 |
22.3 vs 17.4 |
|
BRAF + MEK inhibitors |
COLUMBUS (NCT01909453) |
III |
Untreated BRAFV600E/K melanoma |
Encorafenib+Binimetinib vs Encorafenib vs Vemurafenib |
PFS |
577 |
64 vs 52 vs 41 |
14.9 vs 9.6 vs 6.3 |
33.6 vs 23.5 vs 16.9 |
|
Triplet therapy (ICI+Targeted) |
IMSpire150 (NCT02908672) |
III |
Untreated BRAFV600 melanoma |
Atezo+Vem+Cobi vs Placebo+Vem+Cobi |
PFS |
514 |
66 vs 65 |
15.1 vs 10.0 |
NR |
|
Triplet therapy (ICI+Targeted) |
COMBI-I (NCT02967692) |
III |
Untreated BRAFV600 melanoma |
Spartalizumab+Dab+Tra vs Placebo+Dab+Tra |
PFS |
532 |
69 vs 64 |
16.2 vs 12.0 |
NR |
|
CTLA-4, cytotoxic t lymphocyte associated protein 4; PD-1, programmed death receptor 1; BRAF, the proto-oncogene on human chromosome 7; MEK, methyl ethyl ketone; ICI, immune checkpoint inhibitor; DTIC, dacarbazine; OS, overall survival; PFS, progression-free survival; ORR, objective response rate; NR, not reported. |
|||||||||
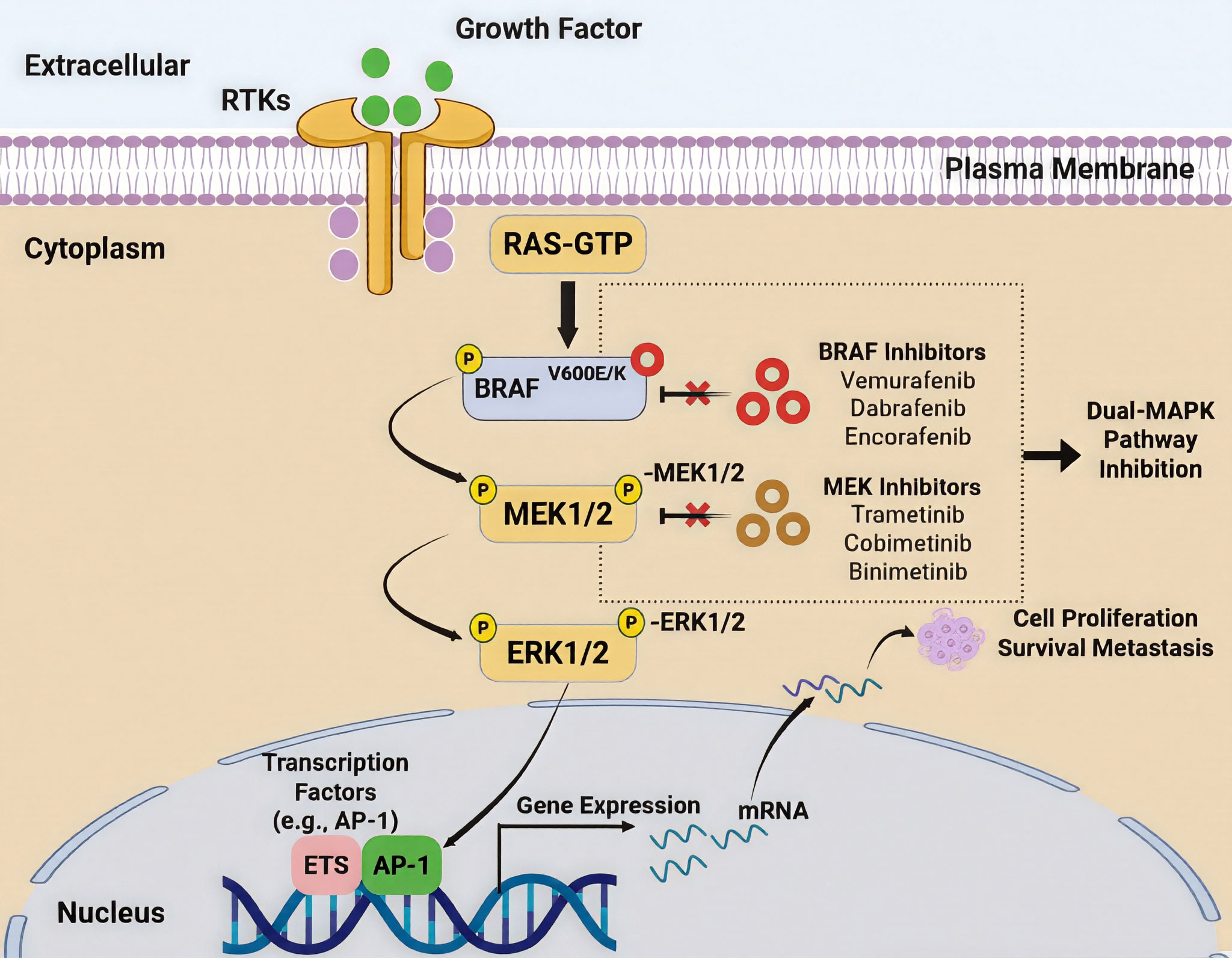 Figure 3. Figure showing the use of the MAPK signaling cascade in BRAF-mutant melanoma as well as its inhibition via pharmacotherapy. RAS is activated in its GTP-bound form by binding of receptor tyrosine kinases (RTKs) on the plasma membrane by extracellular growth factors. Activated RAS activates mutant BRAF (V600E/K), which phosphorylates MEK1/2 and activates it constitutively, resulting in the subsequent phosphorylation of ERK1/2. ERK activation relocates to the nucleus where it controls transcriptional factors like AP-1 and ETS, which support gene expression programs that support cell proliferation, survival, and metastasis. The figure identifies two types of MAPK pathway inhibition with clinically approved BRAF inhibitors (vemurafenib, dabrafenib, encorafenib), MEK inhibitors (trametinib, cobimetinib, binimetinib), which interdicts aberrant signaling at sequential nodes to suppress oncogenic transcriptional outputs and tumor progression.
Figure 3. Figure showing the use of the MAPK signaling cascade in BRAF-mutant melanoma as well as its inhibition via pharmacotherapy. RAS is activated in its GTP-bound form by binding of receptor tyrosine kinases (RTKs) on the plasma membrane by extracellular growth factors. Activated RAS activates mutant BRAF (V600E/K), which phosphorylates MEK1/2 and activates it constitutively, resulting in the subsequent phosphorylation of ERK1/2. ERK activation relocates to the nucleus where it controls transcriptional factors like AP-1 and ETS, which support gene expression programs that support cell proliferation, survival, and metastasis. The figure identifies two types of MAPK pathway inhibition with clinically approved BRAF inhibitors (vemurafenib, dabrafenib, encorafenib), MEK inhibitors (trametinib, cobimetinib, binimetinib), which interdicts aberrant signaling at sequential nodes to suppress oncogenic transcriptional outputs and tumor progression.
|
Table 2. Inhibitors characteristics and their inhibition activity. |
||||
|
Molecular Target / Pathway |
Representative Agents |
Phase |
Investigated Clinical Setting |
Reported ORR (%) |
|
Multi-kinase inhibition (VEGFR1–3, KIT, PDGFR) |
Axitinib |
II / Ib |
Advanced melanoma monotherapy; combined with toripalimab in mucosal melanoma |
18.8; 48.3 |
|
Multi-target TKI (VEGFR, FGFR, KIT, RET) |
Lenvatinib |
I / Ib–II |
Single-agent studies; combination with pembrolizumab in advanced melanoma |
17.2; 48 |
|
KIT-directed inhibition |
Imatinib, Dasatinib Nilotinib |
II |
Evaluated mainly in sun-damaged melanoma mucosal, and acral, subtypes |
23.3-26.2 |
|
IGF-1 receptor blockade |
Linsitinib |
I |
Tested with erlotinib in solid malignancies including melanoma |
1 |
|
EGFR inhibition |
Gefitinib, Erlotinib |
I–II |
Limited activity as monotherapy; explored with PI3K inhibitors in solid tumors |
3.5-4 |
|
VEGF neutralization |
Bevacizumab |
II |
Combined with dacarbazine (cutaneous melanoma) or temozolomide (uveal melanoma) |
18.9; 0 |
|
MEK pathway inhibition |
Pimasertib; Selumetinib |
I–II |
Studied in NRAS-mutant melanoma; compared with temozolomide in advanced disease |
23; 5.8 |
|
PI3K/mTOR dual blockade |
Voxtalisib |
Ib |
Combination trial alongside pimasertib in genetically altered melanoma (limited benefit) |
6 |
|
PI3K inhibition |
Pictilisib |
I |
Evaluated with or alone of erlotinib in solid tumors including melanoma |
3.5–22 |
|
mTOR inhibition |
Everolimus |
I |
Combined with VEGFR inhibitor vatalanib in advanced solid tumors |
12.9 |
|
mTOR inhibition |
Temsirolimus |
II |
Tested with sorafenib; also combined with bevacizumab in BRAF wild-type melanoma |
5; 17.7 |
|
AKT inhibition |
Uprosertib (GSK2141795) |
I |
Combined with trametinib in BRAF wild-type melanoma and other cancers |
<5 |
|
AKT inhibition |
Afuresertib |
I |
Investigated with trametinib in solid tumors and myeloma |
5 |
|
Wnt pathway blockade |
Vantictumab (OMP-18R5) |
Preclinical |
Demonstrated tumor suppression in xenograft models; not yet in melanoma trials |
NA |
|
Wnt secretion inhibitor |
LGK974 |
I |
Recruiting trial as monotherapy or with anti–PD-1 agent (PDR001) |
NA |
|
IKK/NF-κB inhibition |
BMS-345541 |
Preclinical |
Proposed target; no clinical melanoma trials reported |
NA |
|
MITF modulation (HDAC inhibition) |
Panobinostat |
I |
Studied in metastatic melanoma patients |
0 |
|
CDK4/6 inhibition |
Palbociclib |
II / I–II |
Acral melanoma with CDK alterations; combined with vemurafenib in BRAFV600 melanoma |
20; 27.8 |
|
CDK4/6 inhibition |
Abemaciclib |
Preclinical |
Activity observed in BRAF-resistant melanoma models |
NA |
|
NTRK inhibition (next generation) |
Selitrectinib; Repotrectinib |
Preclinical |
Developed for resistance in NTRK/ROS1/ALK fusion cancers |
NA |
|
ALK inhibition |
Ceritinib; Crizotinib |
NA / I |
Sensitive mucosal melanoma models; combination trials in BRAF-mutant tumors |
11; 29 |
|
Spliceosome inhibition (SF3B1) |
E7107 |
I |
Early-phase monotherapy trial in solid tumors |
0 |
|
VEGF, vascular endothelial growth factor; KIT, protooncogene; PDGFR, platelet-derived growth factor receptor; FGFR, fibroblast growth factor receptor; RET, protooncogene; IGF, insulin-like growth factor; EGFR, epidermal growth factor receptor; MEK, methyl ethyl ketone; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; IKK, IκB kinase; NF-kB, Nuclear factor κB; CDK4/6, cyclin-dependent kinases 4/6; NTRK, neurotrophin receptor kinase; ALK, anaplastic lymphoma kinase; SF3B1, protooncogene. |
||||
Recently, the fusion of QSAR with machine learning (ML) and deep learning (DL) systems has sparked a major advancement in the field. Multiple linear regression (or partial least squares)-based classical QSAR models are now being augmented with, recurrent neural networks (RNNs), convolutional neural networks (CNNs), and other AI-driven methods that automatically extract complex structure-activity features in the form of molecular graphs or SMILE strings, and which result in improved predictive accuracy and scalability. Such a deep QSAR paradigm can not only produce nonlinear descriptors-activity relationships but also screen ultra-large compound libraries at high-throughput with increased selectivity and resistance mitigation programs against kinase targets [45, 46].
A recent study that has been relevant and used the machine learning-based QSAR on BRAF inhibitors studied pyrimidine-sulfonamide analogues targeting the BRAF V600E protein. The QSAR model by Srisongkram & Tookkane (2024) [53] utilized support vector regression (SVR) along with 15 molecular fingerprint descriptors to forecast the inhibitory behaviors in a collection of pyrimidine-sulfonamide scaffolds, which form the core structure of most approved BRAF inhibitors. The model performed well in terms of selection, robustness and predictability and found nine major fingerprints which had strong correlation with the inhibition’s activity. These fingerprints were also confirmed by network-based activity cliff analysis and molecular docking and confirmed that they are relevant to binding to the BRAF V600E kinase domain. The authors also used their algorithm as an interactive module of potential screening of new analogues. A second on classical QSAR into larger computational pipelines was of imidazo[2,1-b]oxazole derivatives as potential mutant BRAF kinase inhibitors. Boutalaka et al. (2024) [54] constructed 3D-QSAR models utilizing Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). These standard 3D descriptors integrate steric, electrostatic, hydrophobic, and hydrogen-bonding field contributions. The contour maps obtained identified important structural areas that generated activity against BRAF showing aspects that prefer tighter binding and improved inhibitory properties. In this work the molecular docking and molecular dynamics (MD) simulations were also incorporated to verify binding poses and assess dynamic stability in ATP-binding pocket (PDB: 4G9C). Pharmacokinetic profile and toxicity profile have been evaluated based on the use of ADMET predictions with the identification of several imidazo [2,1-b]oxazole scaffolds exhibiting desirable properties.
Singh et al. (2022) [55] developed a 3D-QSAR models relying on Gaussian fields for pyrimidine-sulfonamide hybrid BRAF inhibitors (V600E), correlating the intensity of steric and electrostatic fields to biological activity and structural understanding to optimize the activity. This theoretical study supported future applications of machine learning through the confirmation of the importance of important substituents in the ATP-binding and the RAF-selective binding pockets. In general melanoma-active compounds QSAR studies outside the narrowly concerned BRAF datasets have also led to a refinement of the methodology. As an example, QSAR using molecular docking of cytotoxic NCI compounds against the SK-MEL- 2 melanoma cell line, including interaction with the V600E-BRAF receptor, showed that a careful change of key aromatic moieties could improve binding affinity relative to such gold standards as vemurafenib. AN2 and AC4 derivatives were capable of docking with better scores in silico, and this is an indication of how QSAR models may help prioritize the modifications needed on lead to proceed with synthesis and testing [56]. In these studies, there are several important methodological aspects. Conventional 2D descriptors (e.g. molecular fingerprints, atom-based features) can still be useful in first SAR mapping, especially in machine learning models where high-dimensional sets of descriptors enhance prediction. Nonlinear regressors like SVR and advanced kernels such as CoMFA and CoMSIA supplement the richness of descriptors by taking into consideration the spatial distributions of the electromagnetic fields as well as the electronic fields, which hold great significance in the description of the complex relationships between functionality groups and activity [57, 58]. QSAR in combination with docking and MD provides more predictive confidence as binding modes and dynamic stability are validated, which is crucial in lead optimization [59].
Irrespective of such developments, there are constraints and challenges. The small size of datasets and the range of chemicals can limit the external predictivity and reliability of applicability domain of many QSAR models. Choosing descriptors and overfitting are the old problems, particularly in the context of complex fingerprints, which are not carefully validated [60]. In addition, ligand-based models in the case of BRAF inhibitors have the potential to not fully recapitulate the effects of conformational flexibility or allosteric modulations by mutations outside the V600E site [61]. Combining structural data with ligand-based QSAR (e.g., hybrid pharmacophore-QSAR models) is potentially effective in overcoming such shortcomings, though more complex alignment techniques are needed and bigger datasets are required. However, the increasing convergence of machine learning, field-based QSAR, and dynamic simulations allows predicting the structure-activity landscape more firmly, and smaller tuning of the impact of substituents on both potency and selectivity. As can be seen by recent work, QSAR may not only suggest synthesis-guiding changes but also hint more effectively at deeper structures like activity cliffs and feature importance landscapes that would not have been revealed by more traditional SAR techniques. Along with the current progress in computing capability and the descriptor algorithm, the optimization of QSAR remains a potent pillar of the rational creation of future-generation BRAF inhibitors to treat melanoma.
The third key aspect of integration is an improvement of AI and machine learning superimposed on QSAR/docking/pharmacophore frameworks. Surveys of computational strategies centered on kinases point to the fact that machine learning-enhanced QSAR models with deep learning-based deep neural networks such as CNNs and RNNs are capable of independently identifying hierarchical and synergistic feature collections and improving selectivity and activity prediction to a significant level over classical linear QSAR. These improved ones frequently use structural databases, descriptor fingerprints, and experimental binding information in a combined training procedure, providing stronger classifiers to the results of kinase inhibition (Shahin et al., 2024). Besides, AI has the ability to quickly rank poses that are docked or pharmacophore hypotheses by learning scoring functions that are more likely to correlate with experimental data, accelerating virtual screening campaigns without compromising accuracy [64].
These combined in silico methods provide a compound lens, as a result of which it is possible to navigate chemical space more efficiently. QSAR adds predictive structure-activity relationships, docking positions the relationships to physical binding landscapes, and pharmacophore models present blueprints of predictable interactions. Such hybrid pipelines, with machine learning as reinforcement, provide multi-objective optimization, balancing potency and selectivity and drug-likeness, and greatly decrease the use of tedious experimental screening. This system’s view is especially useful in cancer immunotherapeutic indicators such as mutant BRAF, in which conformational plasticity, resistance mechanisms, and off-target profile limit the application of single-method design paradigms. Figure 4 described the QSAR integration with other Insilico approaches.
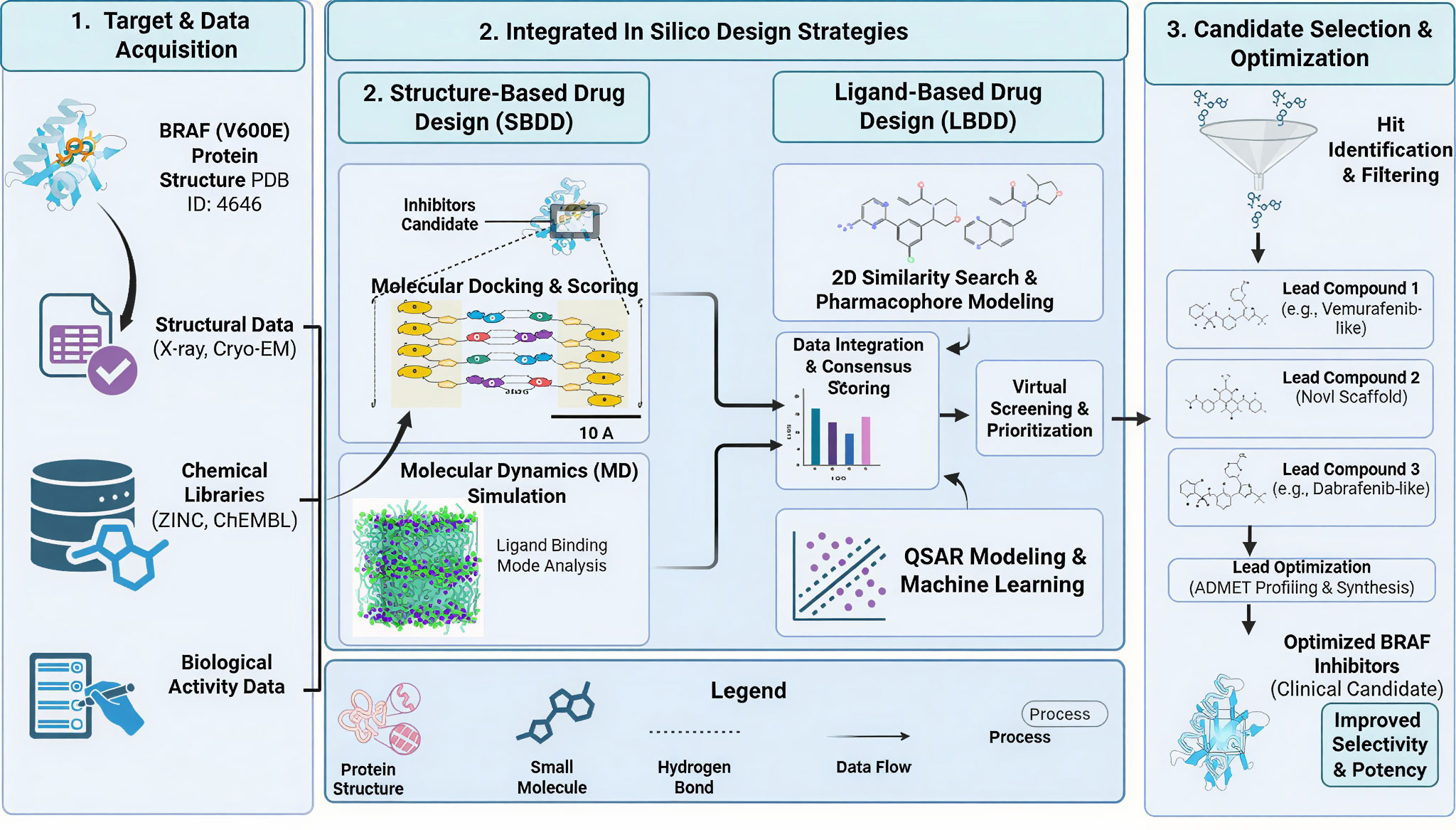 Figure 4. A schematic depiction of an integrated in silico drug discovery pathway to identify and maximize BRAF V600E inhibitors. In Step 1, target and data acquisition, which involves aspect of retrieval of the BRAF V600E protein structure, experimental structural data (X-ray crystallography and cryo-EM), curated chemical libraries (ZINC, ChEMBL), and biological activity datasets, are depicted. Step 2 identifies complementary computational design approaches that combine structural based drug design (SBDD) and ligand based drug design (LBDD). SBDD was performed, and molecular binding modes and stabilizations of the BRAF active site were examined with the help of molecular docking, scoring, and molecular dynamics. LBDD combines similarity searching (2D), pharmacophore modeling, QSAR analysis and machine learning to predict activity and rank compounds. The results of such approaches are combined by consensus scoring and virtual screening. Step 3 demonstrates candidate selection and optimization which encompasses hit identification, lead prioritization, scaffold diversification, and ADMET profiling, which have resulted in optimized BRAF inhibitors with increased selectivity, potency and clinical potential.
Figure 4. A schematic depiction of an integrated in silico drug discovery pathway to identify and maximize BRAF V600E inhibitors. In Step 1, target and data acquisition, which involves aspect of retrieval of the BRAF V600E protein structure, experimental structural data (X-ray crystallography and cryo-EM), curated chemical libraries (ZINC, ChEMBL), and biological activity datasets, are depicted. Step 2 identifies complementary computational design approaches that combine structural based drug design (SBDD) and ligand based drug design (LBDD). SBDD was performed, and molecular binding modes and stabilizations of the BRAF active site were examined with the help of molecular docking, scoring, and molecular dynamics. LBDD combines similarity searching (2D), pharmacophore modeling, QSAR analysis and machine learning to predict activity and rank compounds. The results of such approaches are combined by consensus scoring and virtual screening. Step 3 demonstrates candidate selection and optimization which encompasses hit identification, lead prioritization, scaffold diversification, and ADMET profiling, which have resulted in optimized BRAF inhibitors with increased selectivity, potency and clinical potential.
Model interpretability and transparency is another challenge that has remained. The QSAR algorithms of the past are commonly based on linear descriptors that are readily interpretable, but possess a limited expressiveness, whereas the modern machine learning and deep-learning examples are more predictive but are often treated as black boxes. This obscurity precludes the derivation of mechanistic information and is a nuisance to regulatory acceptance, in which a clear knowledge of how a foretelling was produced is becoming more significant to the safety evaluation and decision-making procedure.
QSAR models also have a limited usefulness as well as applicability domain (AD). The inherent nature of each of the models is that it can only be used to give predictions within the chemical and biological space that the model is trained on, and its extrapolations are speculative and unreliable. The quantification of the AD and its expansion represented by uncertainty estimation and domain metrics is an active field of study but not yet easily applicable in practice [65]. The fast-growing adoption of artificial intelligence (AI) and machine learning processes into the QSAR systems present enormous potential to identify the intricate nonlinear structure-activity relationships and allow multi-objective optimization. This integration does not go without challenges though. The attributes of AI models are generally large high-quality datasets and meticulous feature engineering to avoid biases and overfitting; skewed or incomplete data can distort the learning process and diminish model robustness. Another challenge involves ethical and regulatory reviews, where models aiding drug discovery must demonstrate transparency and validation to gain acceptance by regulators and clinicians.
Going forward, the use of personalized medicine in computational design is both an opportunity and a new complication. Precision medicine seeks to personalize treatment in accordance with personal genomic, proteomic and phenotypic data, which requires QSAR and AI models capable of combining multi-omics data and considering the unique response of a patient to drugs. Although such a transformation can lead to more patient-centric therapeutic design, it adds to the data needs and computational expenses, which highlights the point of federated learning, privacy-preserving algorithms, and interoperable data standards [66]. The next steps will depend on combined activities to enhance data curation and sharing, create explainable AI that interacts between computational predictions and mechanistic understanding and enhance cooperation among computational scientists, medicinal chemists, and clinicians. These challenges can be overcome by QSAR and AI-enhanced modeling to achieve their full potential of facilitating precision drug discovery and personalized therapeutic interventions.
No applicable.
Ethics approval
No applicable.
Data availability
The data will be available upon request.
Funding
None.
Authors’ contribution
XCY, YD, GYZ, ZYC and YYZ performed the literature search and data collection. YWZ, CPL, ZHF, ZHM, ZHM, JX and ZXW conducted the critical analysis of the included studies. XCY, YD, ZXW, WKH, XY, JG and DH wrote the masnuctipt.
Competing interests
The authors declare no competing interests.
- Lo JA, Fisher DE: The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 2014, 346(6212): 945-949.
- Chattopadhyay C, Kim DW, Gombos DS, Oba J, Qin Y, Williams MD, Esmaeli B, Grimm EA, Wargo JA, Woodman SE: Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122(15): 2299-2312.
- Bai J, Wan Z, Zhou W, Wang L, Lou W, Zhang Y, Jin H: Global trends and emerging insights in BRAF and MEK inhibitor resistance in melanoma: a bibliometric analysis. Front Mol Biosci 2025, 12: 1538743.
- Patel H, Yacoub N, Mishra R, White A, Yuan L, Alanazi S, Garrett JT: Current advances in the treatment of BRAF-mutant melanoma. Cancers 2020, 12(2): 482.
- Castellani G, Buccarelli M, Arasi MB, Rossi S, Pisanu ME, Bellenghi M, Lintas C, Tabolacci C: BRAF mutations in melanoma: biological aspects, therapeutic implications, and circulating biomarkers. Cancers 2023, 15(16): 4026.
- Ottaviano M, Giunta EF, Tortora M, Curvietto M, Attademo L, Bosso D, Cardalesi C, Rosanova M, De Placido P, Pietroluongo E: BRAF gene and melanoma: Back to the future. Int J Mol Sci 2021, 22(7): 3474.
- Kong BY, Carlino MS, Menzies AM: Biology and treatment of BRAF mutant metastatic melanoma. Melanoma Manag 2016, 3(1): 33-45.
- Ito T, Kaku-Ito Y, Murata M, Ichiki T, Kuma Y, Tanaka Y, Ide T, Ohno F, Wada-Ohno M, Yamada Y: Intra-and inter-tumor BRAF heterogeneity in acral melanoma: An immunohistochemical analysis. Int J Mol Sci 2019, 20(24): 6191.
- Nepote A, Avallone G, Ribero S, Cavallo F, Roccuzzo G, Mastorino L, Conforti C, Paruzzo L, Poletto S, Schianca FC: Current controversies and challenges on BRAF V600K-mutant cutaneous melanoma. J Clin Med 2022, 11(3): 828.
- Chen G, Gao C, Gao X, Zhang DH, Kuan S-F, Burns TF, Hu J: Wnt/β-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol Cancer Ther 2018, 17(4): 806-813.
- Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, Mina L, Dragovich T, Gimbel M, Mahmoud F: Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther 2018Epub ahead of print.: 7095-7107.
- Anaya YA, Bracho RP, Chauhan SC, Tripathi MK, Bandyopadhyay D: Small Molecule B-RAF inhibitors as anti-cancer therapeutics: Advances in discovery, development, and mechanistic insights. Int J Mol Sci 2025, 26(6): 2676.
- Delord J-P, Robert C, Nyakas M, McArthur GA, Kudchakar R, Mahipal A, Yamada Y, Sullivan R, Arance A, Kefford RF: Phase I dose-escalation and-expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin Cancer Res 2017, 23(18): 5339-5348.
- Cosci I, Salizzato V, Del Fiore P, Pigozzo J, Guarneri V, Mocellin S, Ferlin A, Mathlouthi S, Piccin L, Garofalo M: Molecular basis of BRAF inhibitor resistance in melanoma: a systematic review. Pharmaceuticals 2025, 18(8): 1235.
- Hahn VS, Zhang KW, Sun L, Narayan V, Lenihan DJ, Ky B: Heart failure with targeted cancer therapies: mechanisms and cardioprotection. Circ Res 2021, 128(10): 1576-1593.
- Singh S, Sharma B, Kanwar SS, Kumar A: Lead phytochemicals for anticancer drug development. Front Plant Sci 2016, 7: 1667.
- Arnold M, Singh D, Laversanne M, Vignat J, Vaccarella S, Meheus F, Cust AE, De Vries E, Whiteman DC, Bray F: Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol 2022, 158(5): 495-503.
- Shuvo MSP, Niaz SR, Jannat S, Hossain MA, Mashhur N, Ahmed N, Sohel M, Hasan MI, Ansari SA, Humayoo M: Computational and pharmacophore-based study of Camellia sinensis phytochemicals targeting BRAF in melanoma. Sci Rep 2025, 15(1): 38339.
- Lu Z, Zhang A: AI-driven de novo design of BRAF inhibitors with enhanced binding affinity and optimized drug-likeness. PeerJ 2026, 14: e20541.
- Teixido C, Castillo P, Martinez-Vila C, Arance A, Alos L: Molecular markers and targets in melanoma. Cells 2021, 10(9): 2320.
- Raman M, Chen W, Cobb M: Differential regulation and properties of MAPKs. Oncogene 2007, 26(22): 3100-3112.
- Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP: Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res 2003, 63(11): 2881-2890.
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T: Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22(5): 668-682.
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K: Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010, 363(9): 809-819.
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R: Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018, 19(5): 603-615.
- Chapman P: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2015, 372: 30.
- Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Kaempgen E: Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380(9839): 358-365.
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I: BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439(7074): 358-362.
- Haist M, Stege H, Ebner R, Fleischer MI, Loquai C, Grabbe S: The Role of Treatment Sequencing with Immune-Checkpoint Inhibitors and BRAF/MEK Inhibitors for Response and Survival of Patients with BRAFV600-Mutant Metastatic Melanoma—A Retrospective, Real-World Cohort Study. Cancers 2022, 14(9): 2082.
- Planchard D, Besse B, Groen HJ, Hashemi SM, Mazieres J, Kim TM, Quoix E, Souquet P-J, Barlesi F, Baik C: Phase 2 study of dabrafenib plus trametinib in patients with BRAF V600E-mutant metastatic NSCLC: updated 5-year survival rates and genomic analysis. J Thorac Oncol 2022, 17(1): 103-115.
- Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B: Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PloS one 2011, 6(12): e28973.
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K: Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 2013, 3(3): 338-349.
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB: Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014, 4(1): 80-93.
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G: Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352(6282): 189-196.
- Misek SA, Appleton KM, Dexheimer TS, Lisabeth EM, Lo R, Larsen SD, Gallo K, Neubig RR: Rho-mediated signaling promotes BRAF inhibitor resistance in de-differentiated melanoma cells. Oncogene 2020, 39(7): 1466-1483.
- Teixidó C, González-Cao M, Karachaliou N, Rosell R: Predictive factors for immunotherapy in melanoma. Ann Transl Med 2015, 3(15): 208.
- Fs H: Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010, 363: 711-723.
- Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain J-F, Testori A, Grob J-J: Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011, 364(26): 2517-2526.
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E: Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015, 372(4): 320-330.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M: Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015, 372(26): 2521-2532.
- Hamid O, Robert C, Daud A, Hodi F, Hwu W, Kefford R, Wolchok J, Hersey P, Joseph R, Weber J: Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 2019, 30(4): 582-588.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P: Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015, 373(1): 23-34.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF: Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017, 377(14): 1345-1356.
- Vasilev B, Atanasova M: A (comprehensive) review of the application of quantitative structure–activity relationship (QSAR) in the prediction of new compounds with anti-breast cancer activity. Appl Sci 2025, 15(3): 1206.
- Koirala M, Yan L, Mohamed Z, DiPaola M: AI-Integrated QSAR modeling for enhanced drug discovery: from classical approaches to deep learning and structural insight. Int J Mol Sci 2025, 26(19): 9384.
- Shahin R, Jaafreh S, Azzam Y: Tracking protein kinase targeting advances: integrating QSAR into machine learning for kinase-targeted drug discovery. Future Sci OA 2025, 11(1): 2483631.
- Pei M, Qian A, Cao L, Wang Z, Lu Y, Yan C, Liang T: In silico design of novel potential isonicotinamide-based glycogen synthase kinase-3β (GSK-3β) inhibitors: 3D-QSAR, molecular docking, molecular dynamics simulation and ADMET studies. NJC 2025, 49(15): 6151-6163.
- Faris A, Ibrahim IM, Alnajjar R, Hadni H, Bhat MA, Yaseen M, Chakraborty S, Alsakhen N, Shamkh IM, Mabood F: QSAR-driven screening uncovers and designs novel pyrimidine-4, 6-diamine derivatives as potent JAK3 inhibitors. J Biomol Struct Dyn 2025, 43(2): 757-786.
- Giraldo A, Ruiz D, Caruso M, Mancilla J, Bellomo G: Q2SAR: A Quantum Multiple Kernel Learning Approach for Drug Discovery. arXiv preprint arXiv:250614920 2025Epub ahead of print.
- Niazi SK, Mariam Z: Recent advances in machine-learning-based chemoinformatics: a comprehensive review. Int J Mol Sci 2023, 24(14): 11488.
- Roy K: Advances in QSAR modeling. Applications in pharmaceutical, chemical, food, agricultural and environmental sciences 2017, 555: 39.
- De P, Kar S, Ambure P, Roy K: Prediction reliability of QSAR models: an overview of various validation tools. Arch Toxicol 2022, 96(5): 1279-1295.
- Srisongkram T, Tookkane D: Insights into the structure-activity relationship of pyrimidine-sulfonamide analogues for targeting BRAF V600E protein. Biophys Chem 2024, 307: 107179.
- Boutalaka M, El Bahi S, Alaqarbeh M, El Alaouy MA, Koubi Y, Khatabi KE, Maghat H, Bouachrine M, Lakhlifi T: Computational investigation of imidazo [2, 1-b] oxazole derivatives as potential mutant BRAF kinase inhibitors: 3D-QSAR, molecular docking, molecular dynamics simulation, and ADMETox studies. J Biomol Struct Dyn 2024, 42(10): 5268-5287.
- Singh AK, Novak J, Kumar A, Singh H, Thareja S, Pathak P, Grishina M, Verma A, Yadav JP, Khalilullah H: Gaussian field-based 3D-QSAR and molecular simulation studies to design potent pyrimidine–sulfonamide hybrids as selective BRAF V600E inhibitors. RSC Adv 2022, 12(46): 30181-30200.
- Umar AB, Uzairu A, Shallangwa GA, Uba S: QSAR modelling and molecular docking studies for anti-cancer compounds against melanoma cell line SK-MEL-2. Heliyon 2020, 6(3).
- Bahia MS, Kaspi O, Touitou M, Binayev I, Dhail S, Spiegel J, Khazanov N, Yosipof A, Senderowitz H: A comparison between 2D and 3D descriptors in QSAR modeling based on bio‐active conformations. Mol Inform 2023, 42(4): 2200186.
- Shi Y: Support vector regression-based QSAR models for prediction of antioxidant activity of phenolic compounds. Sci Rep 2021, 11(1): 8806.
- Ashraf N, Asari A, Yousaf N, Ahmad M, Ahmed M, Faisal A, Saleem M, Muddassar M: Combined 3D-QSAR, molecular docking and dynamics simulations studies to model and design TTK inhibitors. Front Chem 2022, 10: 1003816.
- Neira-Albornoz A, Martínez-Parga-Méndez M, González M, Spitz A: Understanding requirements, limitations and applicability of QSAR and PTF models for predicting sorption of pollutants on soils: a systematic review. Front Environ Sci 2024, 12: 1379283.
- Kim KH: Outliers in SAR and QSAR: 4. effects of allosteric protein–ligand interactions on the classical quantitative structure–activity relationships. Mol Divers 2022, 26(6): 3057-3092.
- Khedraoui M, Abchir O, Nour H, Yamari I, Errougui A, Samadi A, Chtita S: An in silico study based on QSAR and molecular docking and molecular dynamics simulation for the discovery of novel potent inhibitor against AChE. Pharmaceuticals (Basel) 2024, 17(7): 830.
- Ganji M, Bakhshi S, Shoari A, Ahangari Cohan R: Discovery of potential FGFR3 inhibitors via QSAR, pharmacophore modeling, virtual screening and molecular docking studies against bladder cancer. J Transl Med 2023, 21(1): 111.
- Cieślak M, Danel T, Krzysztyńska-Kuleta O, Kalinowska-Tłuścik J: Machine learning accelerates pharmacophore-based virtual screening of MAO inhibitors. Sci Rep 2024, 14(1): 8228.
- Mora JR, Marquez EA, Pérez-Pérez N, Contreras-Torres E, Perez-Castillo Y, Agüero-Chapin G, Martinez-Rios F, Marrero-Ponce Y, Barigye SJ: Rethinking the applicability domain analysis in QSAR models. J Comput Aided Mol Des 2024, 38(1): 9.
- Serrano DR, Luciano FC, Anaya BJ, Ongoren B, Kara A, Molina G, Ramirez BI, Sánchez-Guirales SA, Simon JA, Tomietto G: Artificial intelligence (AI) applications in drug discovery and drug delivery: Revolutionizing personalized medicine. Pharmaceutics 2024, 16(10): 1328.
Asia-Pacific Journal of Oncology
print ISSN: 2708-7980, online ISSN: 2708-7999
 Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Asia Pac J Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.